

The T=1 capsid protein of Penicillium chrysogenum virus is formed by a repeated helix-rich core indicative of gene duplication
- PMID: 20463071
- PMCID: PMC2898261
- DOI: 10.1128/JVI.00432-10
The T=1 capsid protein of Penicillium chrysogenum virus is formed by a repeated helix-rich core indicative of gene duplication
Abstract
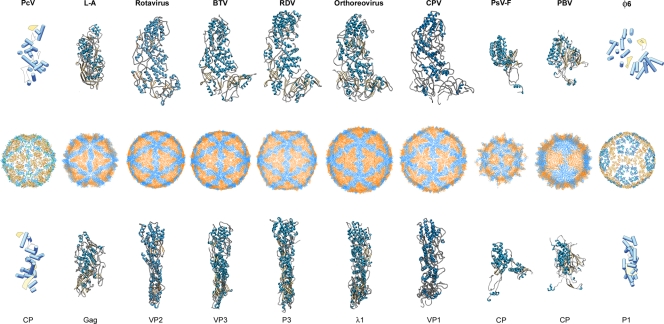
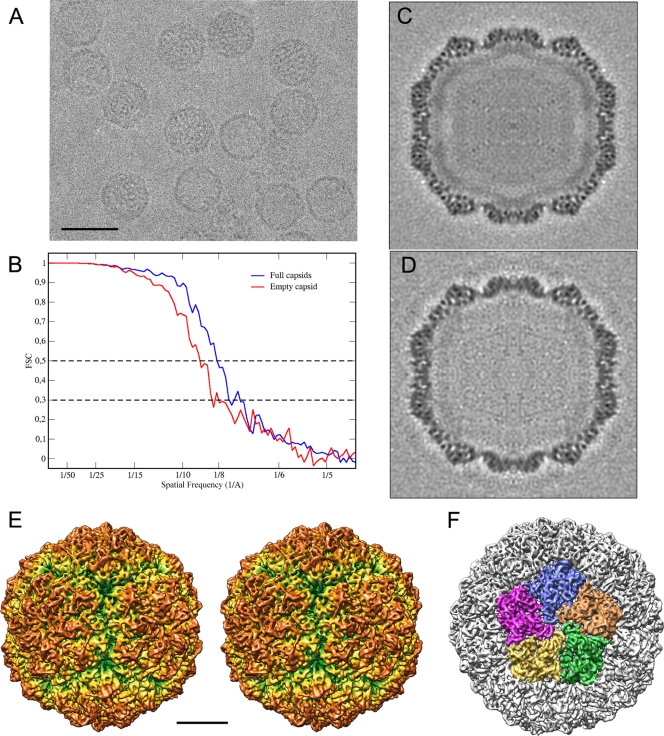
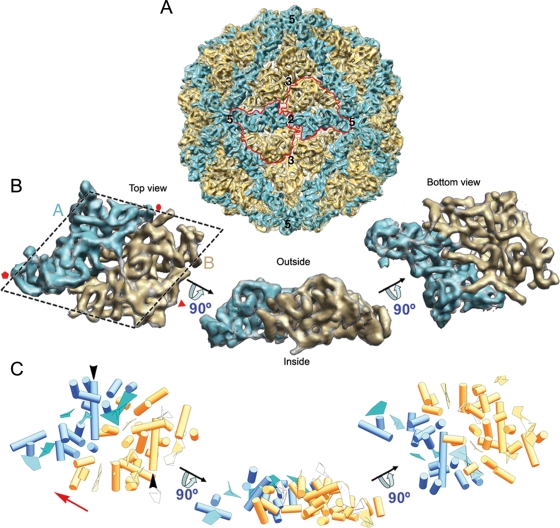
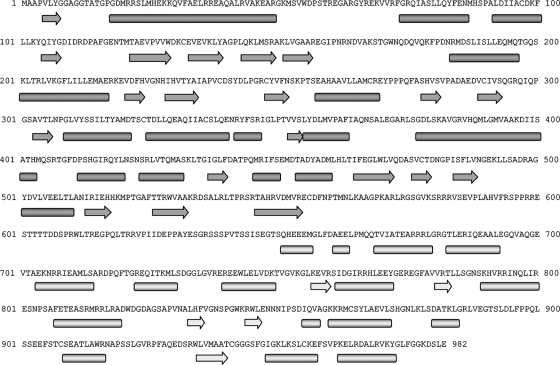
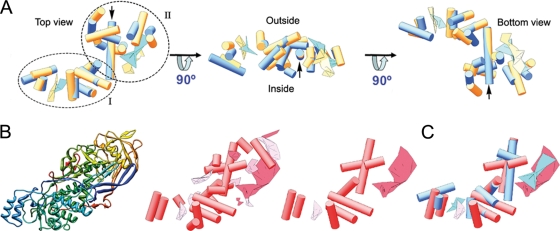
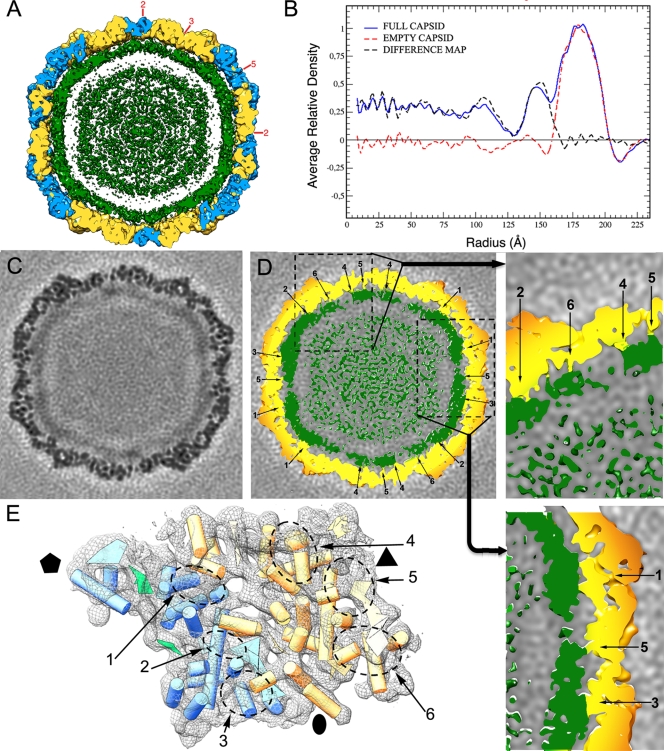
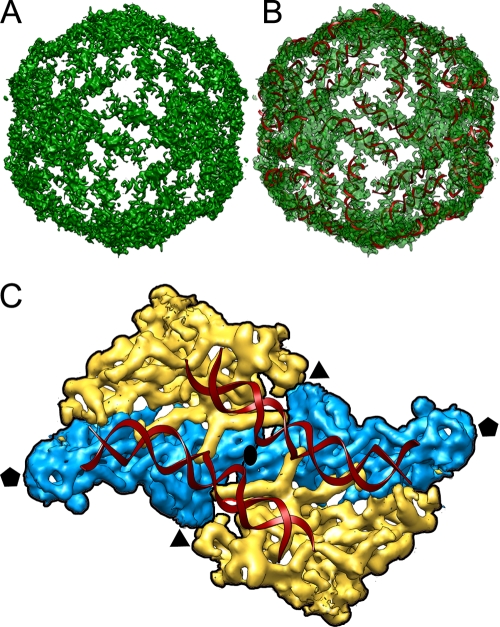
Penicillium chrysogenum virus (PcV), a member of the Chrysoviridae family, is a double-stranded RNA (dsRNA) fungal virus with a multipartite genome, with each RNA molecule encapsidated in a separate particle. Chrysoviruses lack an extracellular route and are transmitted during sporogenesis and cell fusion. The PcV capsid, based on a T=1 lattice containing 60 subunits of the 982-amino-acid capsid protein, remains structurally undisturbed throughout the viral cycle, participates in genome metabolism, and isolates the virus genome from host defense mechanisms. Using three-dimensional cryoelectron microscopy, we determined the structure of the PcV virion at 8.0 A resolution. The capsid protein has a high content of rod-like densities characteristic of alpha-helices, forming a repeated alpha-helical core indicative of gene duplication. Whereas the PcV capsid protein has two motifs with the same fold, most dsRNA virus capsid subunits consist of dimers of a single protein with similar folds. The spatial arrangement of the alpha-helical core resembles that found in the capsid protein of the L-A virus, a fungal totivirus with an undivided genome, suggesting a conserved basic fold. The encapsidated genome is organized in concentric shells; whereas the inner dsRNA shells are well defined, the outermost layer is dense due to numerous interactions with the inner capsid surface, specifically, six interacting areas per monomer. The outermost genome layer is arranged in an icosahedral cage, sufficiently well ordered to allow for modeling of an A-form dsRNA. The genome ordering might constitute a framework for dsRNA transcription at the capsid interior and/or have a structural role for capsid stability.
Figures
References
-
- Abrescia, N. G., J. J. Cockburn, J. M. Grimes, G. C. Sutton, J. M. Diprose, S. J. Butcher, S. D. Fuller, C. San Martin, R. M. Burnett, D. I. Stuart, D. H. Bamford, and J. K. Bamford. 2004. Insights into assembly from structural analysis of bacteriophage PRD1. Nature 432:68-74. - PubMed
-
- Adamczak, R., A. Porollo, and J. Meller. 2005. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins 59:467-475. - PubMed
-
- Agirrezabala, X., J. Martin-Benito, M. Valle, J. M. Gonzalez, A. Valencia, J. M. Valpuesta, and J. L. Carrascosa. 2005. Structure of the connector of bacteriophage T7 at 8A resolution: structural homologies of a basic component of a DNA translocating machinery. J. Mol. Biol. 347:895-902. - PubMed
