

Systemic mitochondrial dysfunction and the etiology of Alzheimer's disease and down syndrome dementia
- PMID: 20463402
- PMCID: PMC4175722
- DOI: 10.3233/JAD-2010-100351
Systemic mitochondrial dysfunction and the etiology of Alzheimer's disease and down syndrome dementia
Abstract
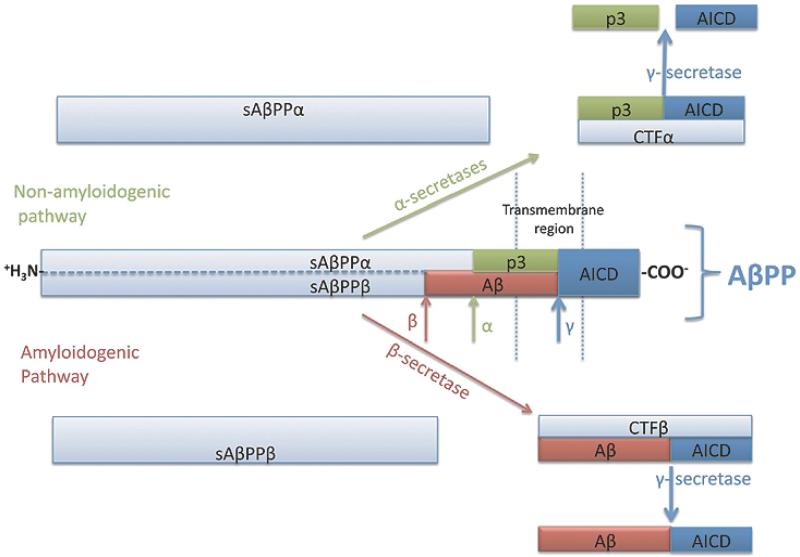
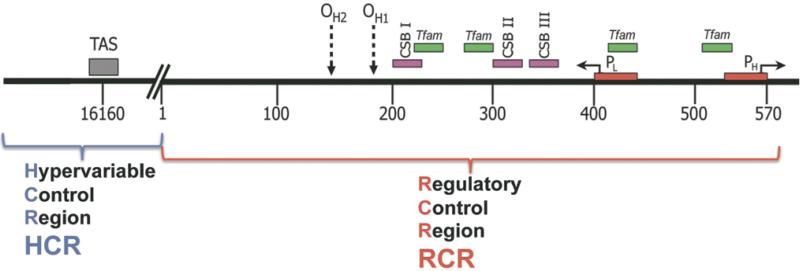
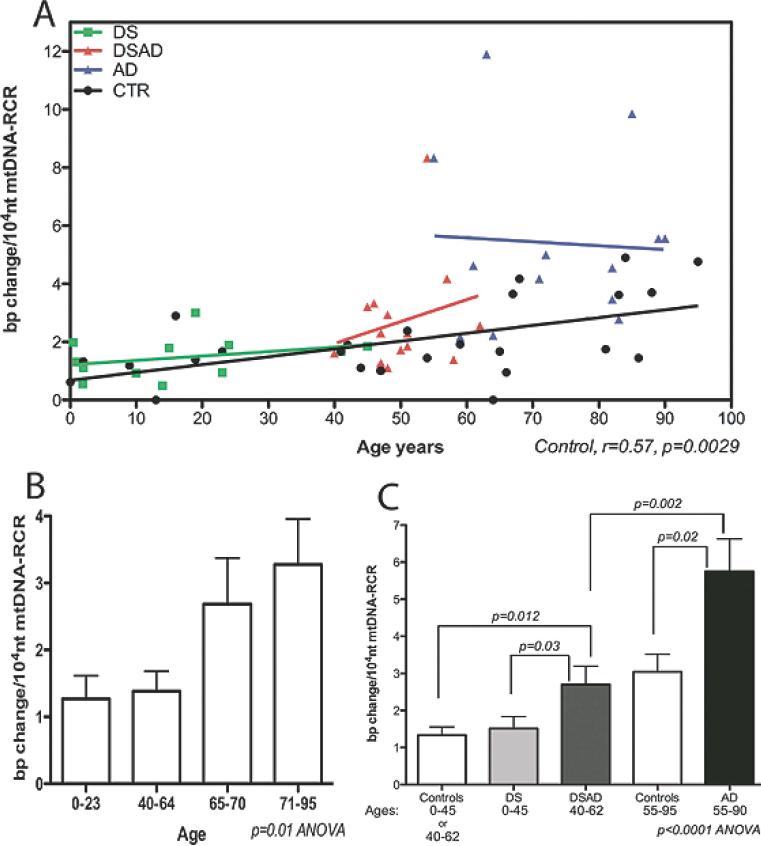
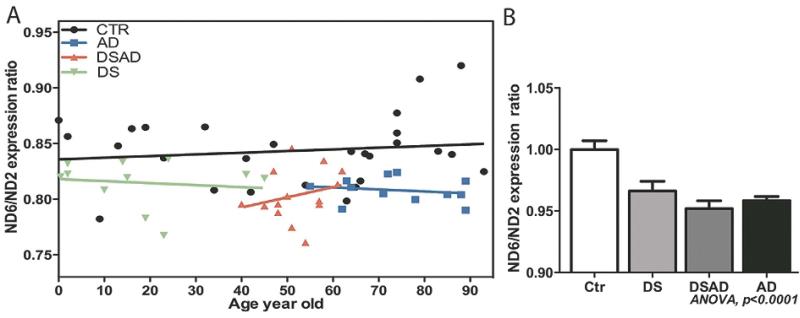
Increasing evidence is implicating mitochondrial dysfunction as a central factor in the etiology of Alzheimer's disease (AD). The most significant risk factor in AD is advanced age and an important neuropathological correlate of AD is the deposition of amyloid-beta peptide (Abeta40 and Abeta42) in the brain. An AD-like dementia is also common in older individuals with Down syndrome (DS), though with a much earlier onset. We have shown that somatic mitochondrial DNA (mtDNA) control region (CR) mutations accumulate with age in post-mitotic tissues including the brain and that the level of mtDNA mutations is markedly elevated in the brains of AD patients. The elevated mtDNA CR mutations in AD brains are associated with a reduction in the mtDNA copy number and in the mtDNA L-strand transcript levels. We now show that mtDNA CR mutations increase with age in control brains; that they are markedly elevated in the brains of AD and DS and dementia (DSAD) patients; and that the increased mtDNA CR mutation rate in DSAD brains is associated with reduced mtDNA copy number and L-strand transcripts. The increased mtDNA CR mutation rate is also seen in peripheral blood DNA and in lymphoblastoid cell DNAs of AD and DSAD patients, and distinctive somatic mtDNA mutations, often at high heteroplasmy levels, are seen in AD and DSAD brain and blood cells DNA. In aging, DS, and DSAD, the mtDNA mutation level is positively correlated with beta-secretase activity and mtDNA copy number is inversely correlated with insoluble Abeta40 and Abeta42 levels. Therefore, mtDNA alterations may be responsible for both age-related dementia and the associated neuropathological changes observed in AD and DSAD.
Figures



References
-
- Selkoe DJ. Alzheimer's disease. Missense on the membrane. Nature. 1995;375:734–735. - PubMed
-
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, Mullan M. Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. - PubMed
-
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. - PubMed
-
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. - PubMed
-
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St. George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
