

DNA methylome in human CD4+ T cells identifies transcriptionally repressive and non-repressive methylation peaks
- PMID: 20463746
- PMCID: PMC2948060
- DOI: 10.1038/gene.2010.24
DNA methylome in human CD4+ T cells identifies transcriptionally repressive and non-repressive methylation peaks
Abstract
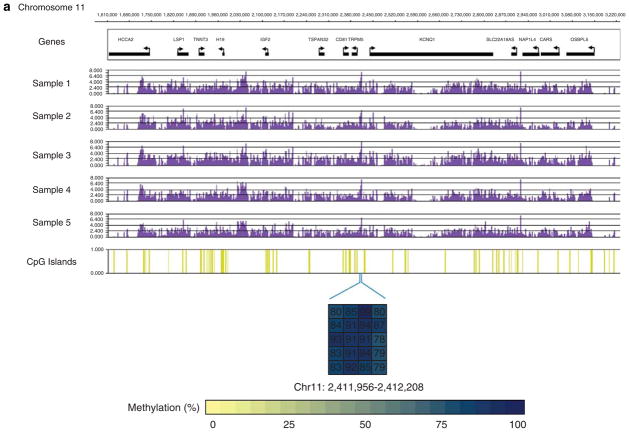
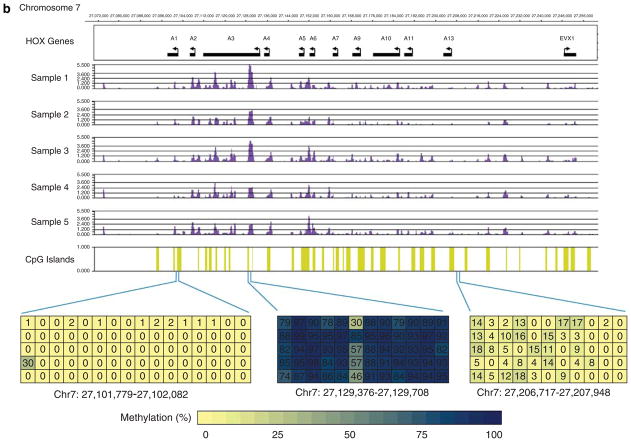
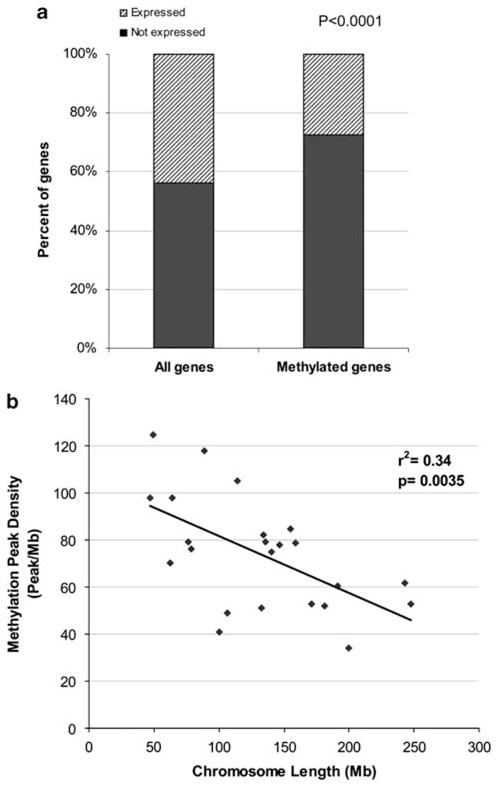
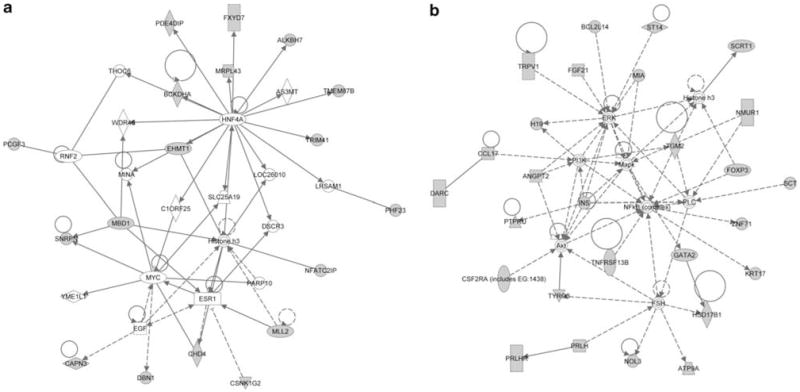
DNA methylation is an epigenetic mark that is critical in determining chromatin accessibility and regulating gene expression. This epigenetic mechanism has an important role in T-cell function. We used genome-wide methylation profiling to characterize the DNA methylome in primary human CD4+ T cells. We found that only 5% of CpG islands are methylated in CD4+ T cells, and that DNA methylation peak density is increased in subtelomeric chromosomal regions. We also found an inverse relationship between methylation peak density and chromosomal length. Our data indicate that DNA methylation in gene promoter regions is not always a repressive epigenetic mark. Indeed, about 27% of methylated genes are actively expressed in CD4+ T cells. We demonstrate that repressive methylation peaks are located closer to the transcription start site (TSS) compared with functionally non-repressive peaks (-893±110 bp versus -1342±218 bp (mean±s.e.m.), P-value <0.05). We also show that both a larger number and an increased CpG island density in promoter sequences predict transcriptional permissiveness of DNA methylation. TSS in the majority of genes with permissive DNA methylation peaks is in DNase I hypersensitive sites, indicating a failure of DNA methylation to induce chromatin inaccessibility in these loci.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science (New York, NY) 2001;293:1089–1093. - PubMed
-
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. - PubMed
-
- Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science (New York, NY) 1981;211:393–396. - PubMed
-
- Bird AP. Functions for DNA methylation in vertebrates. Cold Spring Harb Symp Quant Biol. 1993;58:281–285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
