

Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing
- PMID: 20463816
- PMCID: PMC2865531
- DOI: 10.1371/journal.ppat.1000896
Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing
Abstract
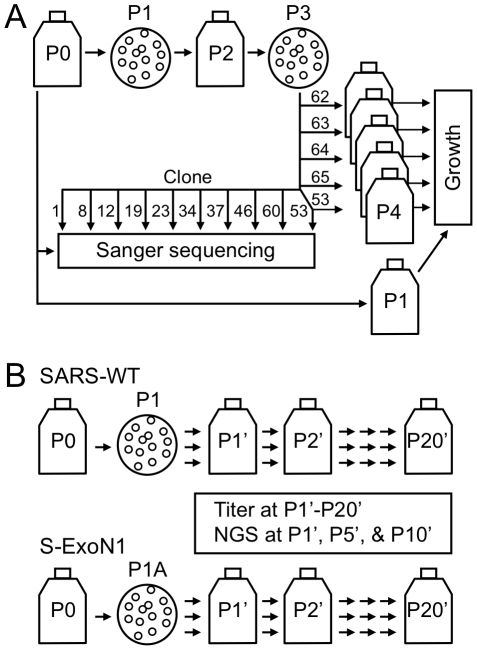
Most RNA viruses lack the mechanisms to recognize and correct mutations that arise during genome replication, resulting in quasispecies diversity that is required for pathogenesis and adaptation. However, it is not known how viruses encoding large viral RNA genomes such as the Coronaviridae (26 to 32 kb) balance the requirements for genome stability and quasispecies diversity. Further, the limits of replication infidelity during replication of large RNA genomes and how decreased fidelity impacts virus fitness over time are not known. Our previous work demonstrated that genetic inactivation of the coronavirus exoribonuclease (ExoN) in nonstructural protein 14 (nsp14) of murine hepatitis virus results in a 15-fold decrease in replication fidelity. However, it is not known whether nsp14-ExoN is required for replication fidelity of all coronaviruses, nor the impact of decreased fidelity on genome diversity and fitness during replication and passage. We report here the engineering and recovery of nsp14-ExoN mutant viruses of severe acute respiratory syndrome coronavirus (SARS-CoV) that have stable growth defects and demonstrate a 21-fold increase in mutation frequency during replication in culture. Analysis of complete genome sequences from SARS-ExoN mutant viral clones revealed unique mutation sets in every genome examined from the same round of replication and a total of 100 unique mutations across the genome. Using novel bioinformatic tools and deep sequencing across the full-length genome following 10 population passages in vitro, we demonstrate retention of ExoN mutations and continued increased diversity and mutational load compared to wild-type SARS-CoV. The results define a novel genetic and bioinformatics model for introduction and identification of multi-allelic mutations in replication competent viruses that will be powerful tools for testing the effects of decreased fidelity and increased quasispecies diversity on viral replication, pathogenesis, and evolution.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Bull JJ, Meyers LA, Lachmann M. Quasispecies made simple. e61PLoS Comput Biol. 2005;1 doi: 10.1371/journal.pcbi.0010061. - DOI - PMC - PubMed
-
- Domingo E, Sabo D, Taniguchi T, Weissmann C. Nucleotide sequence heterogeneity of an RNA phage population. Cell. 1978;13:735–744. - PubMed
-
- Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. e11PLoS Pathog. 2005;1 doi: 10.1371/journal.ppat.0010011. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 AI059443/AI/NIAID NIH HHS/United States
- R01-AI026603/AI/NIAID NIH HHS/United States
- HHSN272200900007C/AI/NIAID NIH HHS/United States
- T32-AI049824/AI/NIAID NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- U54 AI057157/AI/NIAID NIH HHS/United States
- F32-AI080148/AI/NIAID NIH HHS/United States
- R01 AI026603/AI/NIAID NIH HHS/United States
- T32 AI049824/AI/NIAID NIH HHS/United States
- P01-AI059443/AI/NIAID NIH HHS/United States
- F32 AI080148/AI/NIAID NIH HHS/United States
- CA68485/CA/NCI NIH HHS/United States
- U54-AI057157/AI/NIAID NIH HHS/United States
- N01 AI030071/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
