

A differentiation-based phylogeny of cancer subtypes
- PMID: 20463876
- PMCID: PMC2865519
- DOI: 10.1371/journal.pcbi.1000777
A differentiation-based phylogeny of cancer subtypes
Abstract
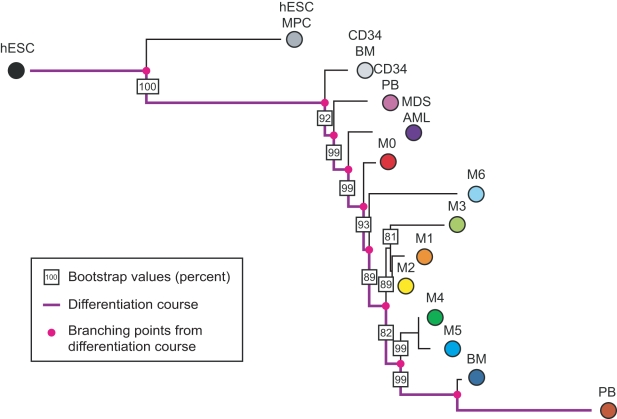
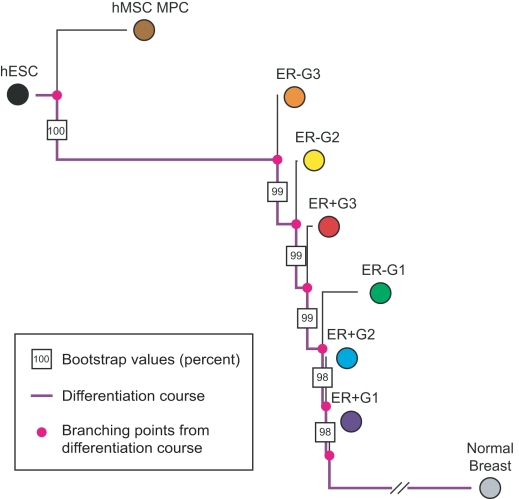
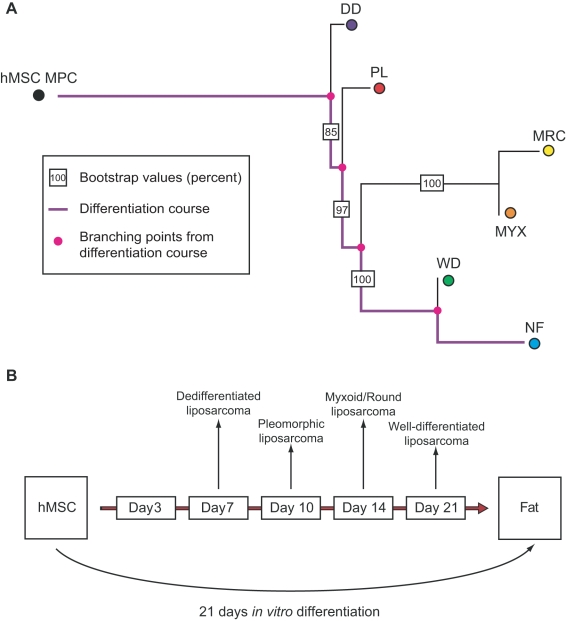
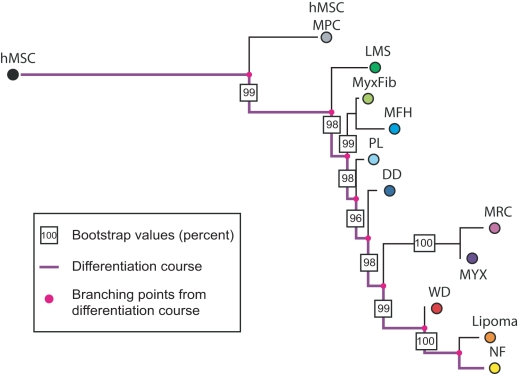
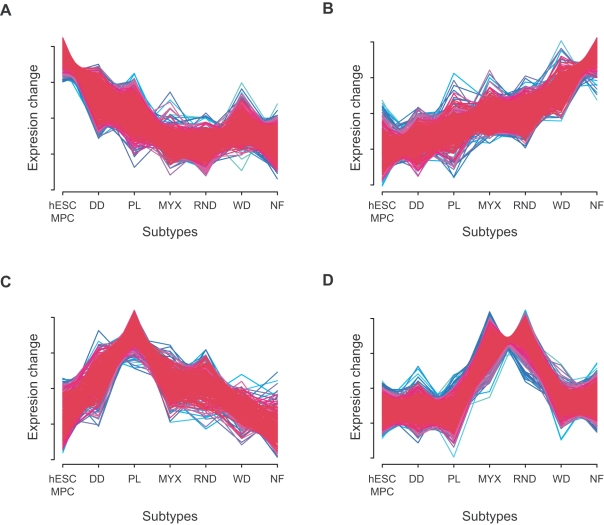
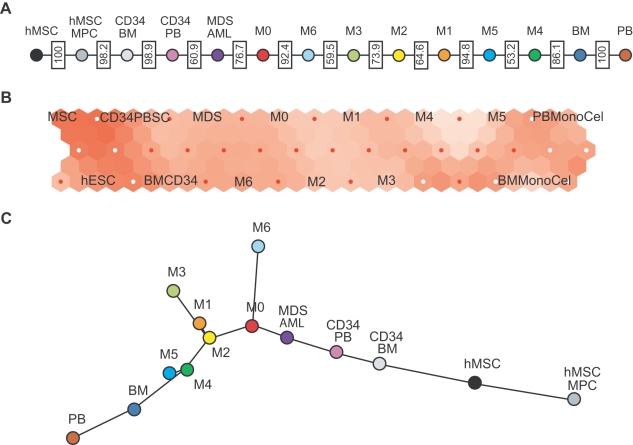
Histopathological classification of human tumors relies in part on the degree of differentiation of the tumor sample. To date, there is no objective systematic method to categorize tumor subtypes by maturation. In this paper, we introduce a novel computational algorithm to rank tumor subtypes according to the dissimilarity of their gene expression from that of stem cells and fully differentiated tissue, and thereby construct a phylogenetic tree of cancer. We validate our methodology with expression data of leukemia, breast cancer and liposarcoma subtypes and then apply it to a broader group of sarcomas. This ranking of tumor subtypes resulting from the application of our methodology allows the identification of genes correlated with differentiation and may help to identify novel therapeutic targets. Our algorithm represents the first phylogeny-based tool to analyze the differentiation status of human tumors.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas.Am J Pathol. 2008 Apr;172(4):1069-80. doi: 10.2353/ajpath.2008.070284. Epub 2008 Feb 29. Am J Pathol. 2008. PMID: 18310505 Free PMC article.
-
A unified computational model for revealing and predicting subtle subtypes of cancers.BMC Bioinformatics. 2012 May 1;13:70. doi: 10.1186/1471-2105-13-70. BMC Bioinformatics. 2012. PMID: 22548981 Free PMC article.
-
Tumor classification ranking from microarray data.BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S21. doi: 10.1186/1471-2164-9-S2-S21. BMC Genomics. 2008. PMID: 18831787 Free PMC article.
-
Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma.Cancer Res. 2007 Jul 15;67(14):6626-36. doi: 10.1158/0008-5472.CAN-07-0584. Cancer Res. 2007. PMID: 17638873
-
MicroRNAs and cancer-the search begins!IEEE Trans Inf Technol Biomed. 2009 Jan;13(1):67-77. doi: 10.1109/TITB.2008.2007086. IEEE Trans Inf Technol Biomed. 2009. PMID: 19129025 Review.
Cited by
-
PhyloOncology: Understanding cancer through phylogenetic analysis.Biochim Biophys Acta Rev Cancer. 2017 Apr;1867(2):101-108. doi: 10.1016/j.bbcan.2016.10.006. Epub 2016 Oct 31. Biochim Biophys Acta Rev Cancer. 2017. PMID: 27810337 Free PMC article. Review.
-
Degree and centrality-based approaches in network-based variable selection: Insights from the Singapore Longitudinal Aging Study.PLoS One. 2019 Jul 18;14(7):e0219186. doi: 10.1371/journal.pone.0219186. eCollection 2019. PLoS One. 2019. PMID: 31318894 Free PMC article.
-
Neural Network Deconvolution Method for Resolving Pathway-Level Progression of Tumor Clonal Expression Programs With Application to Breast Cancer Brain Metastases.Front Physiol. 2020 Sep 4;11:1055. doi: 10.3389/fphys.2020.01055. eCollection 2020. Front Physiol. 2020. PMID: 33013452 Free PMC article.
-
Robust and accurate deconvolution of tumor populations uncovers evolutionary mechanisms of breast cancer metastasis.Bioinformatics. 2020 Jul 1;36(Suppl_1):i407-i416. doi: 10.1093/bioinformatics/btaa396. Bioinformatics. 2020. PMID: 32657393 Free PMC article.
-
Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes.Nat Commun. 2018 Jul 17;9(1):2759. doi: 10.1038/s41467-018-05134-3. Nat Commun. 2018. PMID: 30018341 Free PMC article.
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–458. - PubMed
-
- Kooby DA, Antonescu CR, Brennan MF, Singer S. Atypical lipomatous tumor/well-differentiated liposarcoma of the extremity and trunk wall: importance of histological subtype with treatment recommendations. Ann Surg Oncol. 2004;11:78–84. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
