

Sequence embedding for fast construction of guide trees for multiple sequence alignment
- PMID: 20470396
- PMCID: PMC2893182
- DOI: 10.1186/1748-7188-5-21
Sequence embedding for fast construction of guide trees for multiple sequence alignment
Abstract
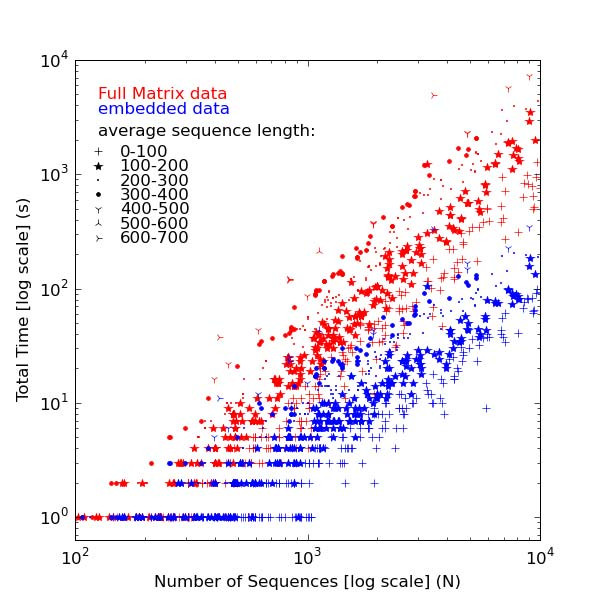
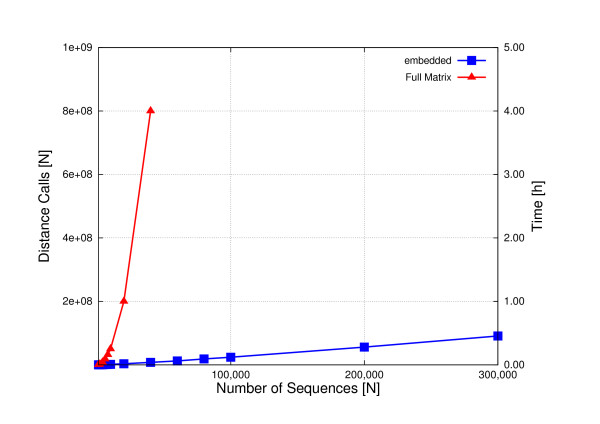
Background: The most widely used multiple sequence alignment methods require sequences to be clustered as an initial step. Most sequence clustering methods require a full distance matrix to be computed between all pairs of sequences. This requires memory and time proportional to N2 for N sequences. When N grows larger than 10,000 or so, this becomes increasingly prohibitive and can form a significant barrier to carrying out very large multiple alignments.
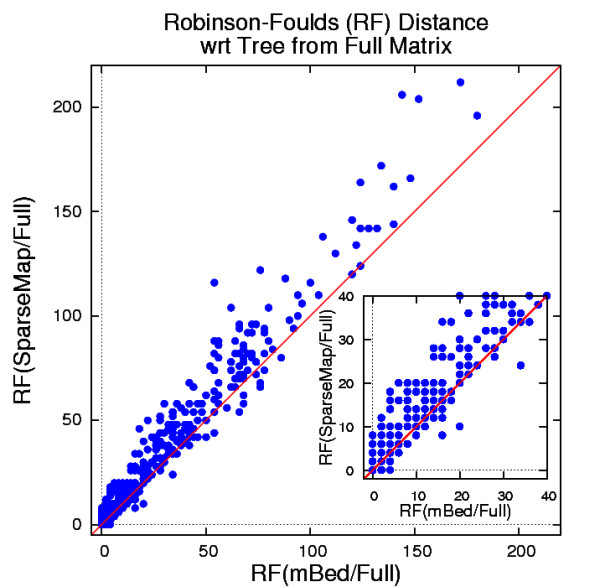
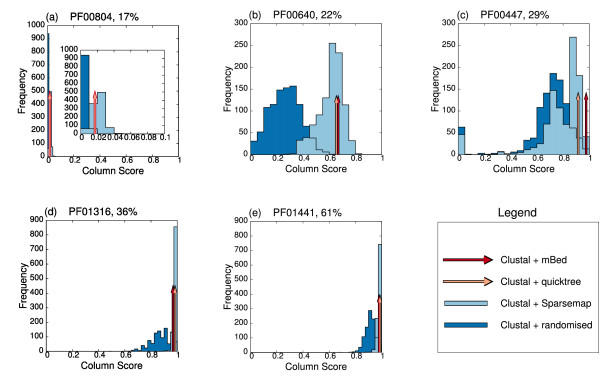
Results: In this paper, we have tested variations on a class of embedding methods that have been designed for clustering large numbers of complex objects where the individual distance calculations are expensive. These methods involve embedding the sequences in a space where the similarities within a set of sequences can be closely approximated without having to compute all pair-wise distances.
Conclusions: We show how this approach greatly reduces computation time and memory requirements for clustering large numbers of sequences and demonstrate the quality of the clusterings by benchmarking them as guide trees for multiple alignment. Source code is available for download from http://www.clustal.org/mbed.tgz.
Figures
References
-
- Taylor WR. Multiple sequence alignment by a pairwise algorithm. Comput Appl Biosci. 1987;3(2):81–7. - PubMed
LinkOut - more resources
Full Text Sources
