

Analysis of the complete genome sequences of human rhinovirus
- PMID: 20471068
- PMCID: PMC2893015
- DOI: 10.1016/j.jaci.2010.04.010
Analysis of the complete genome sequences of human rhinovirus
Abstract
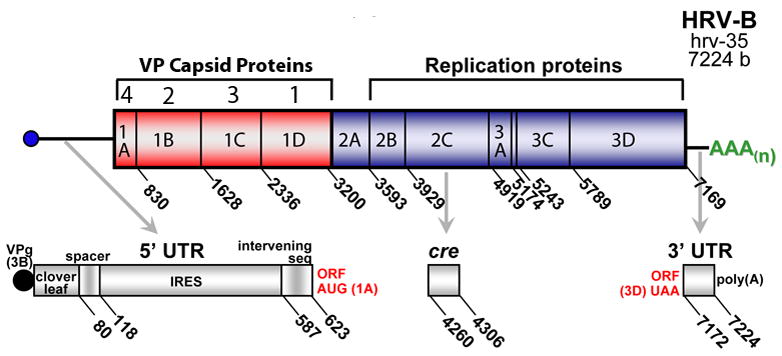
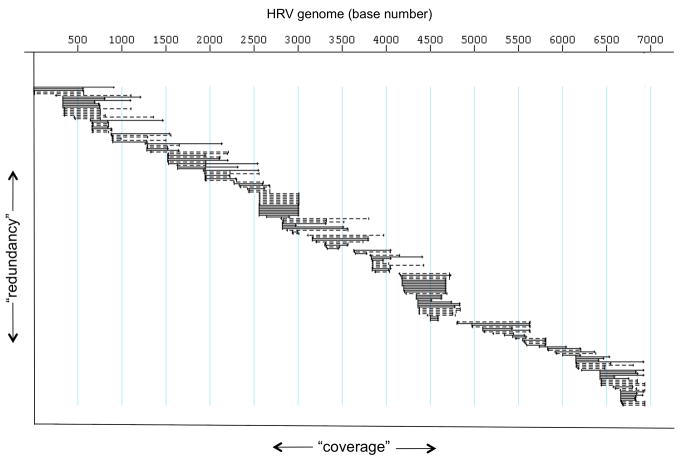
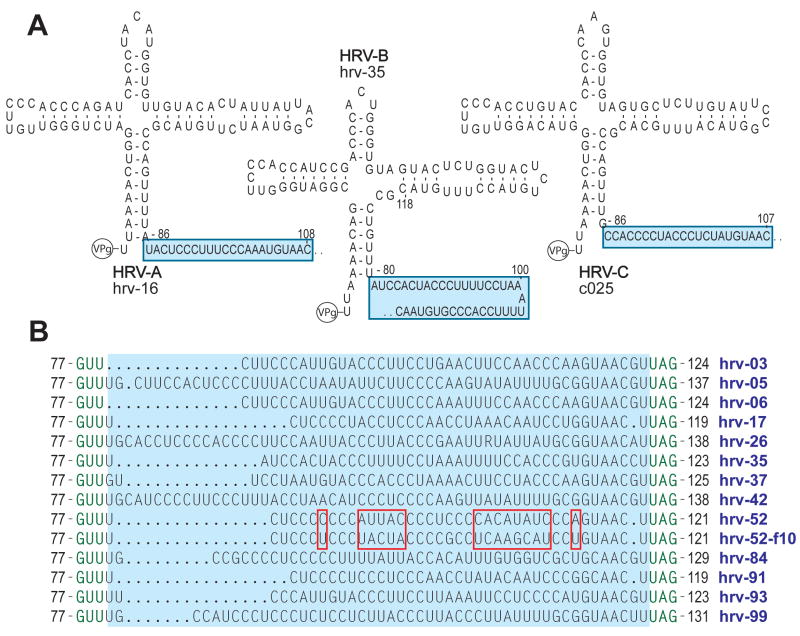
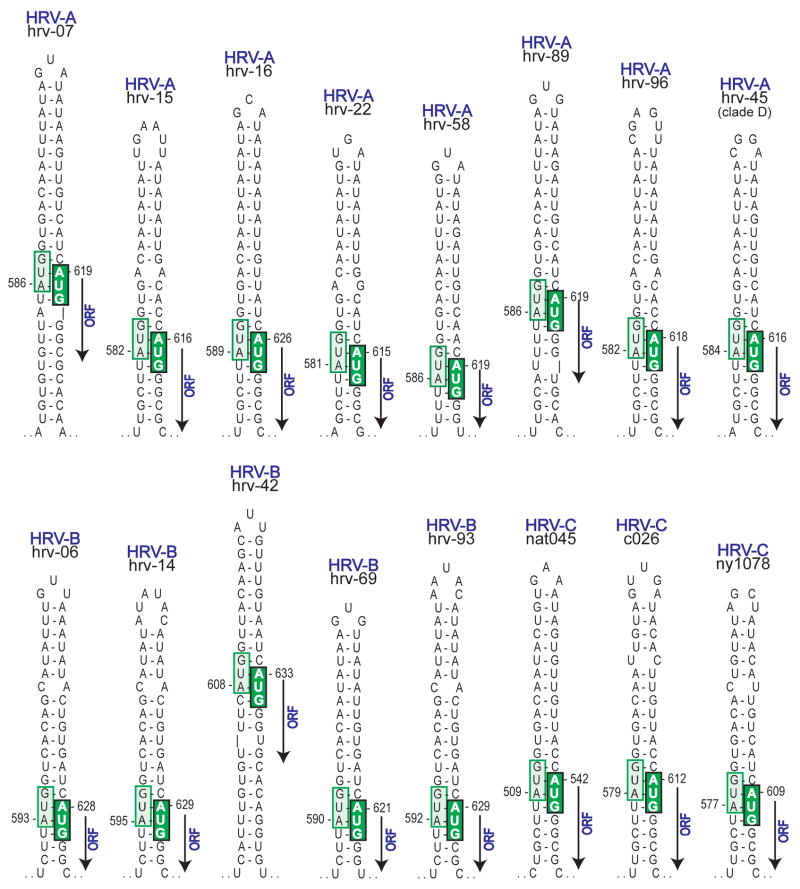
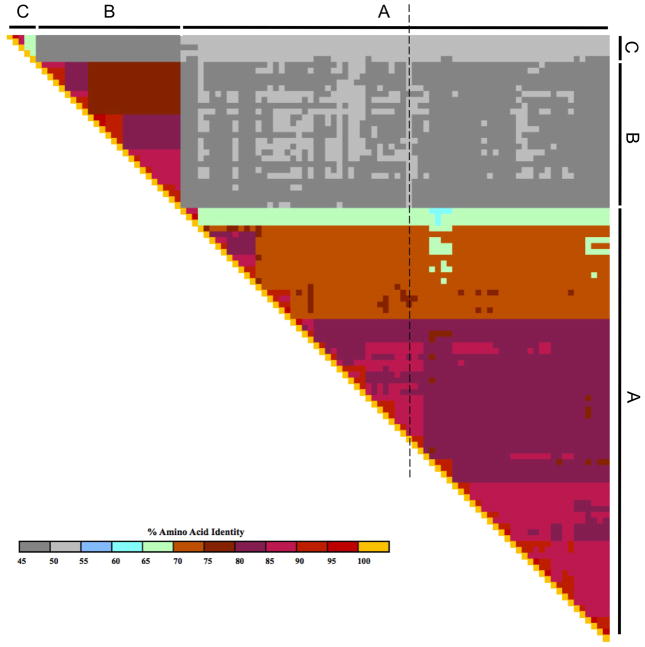
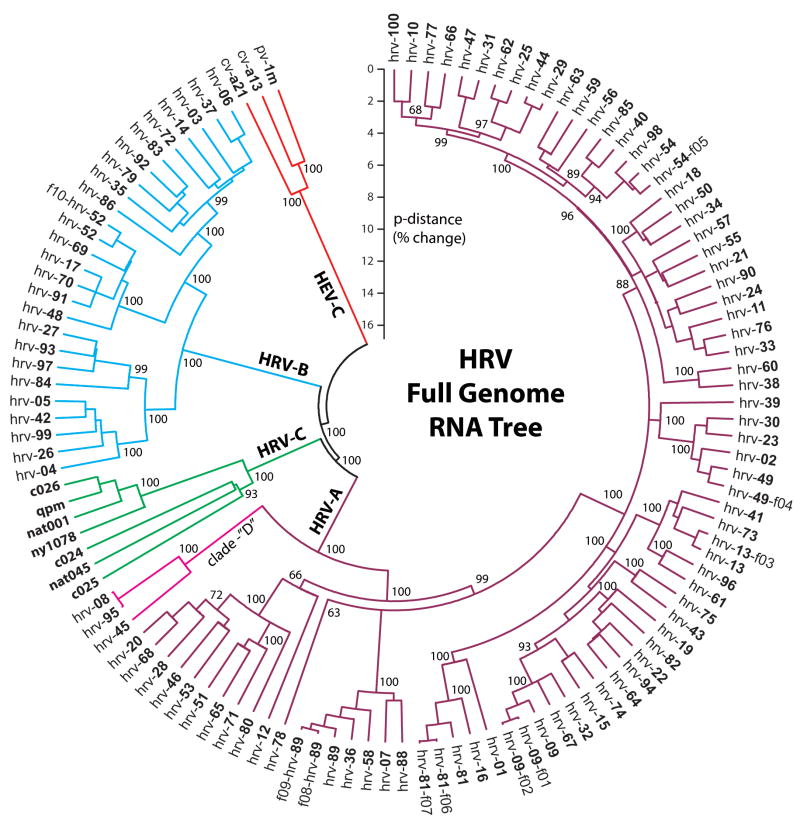
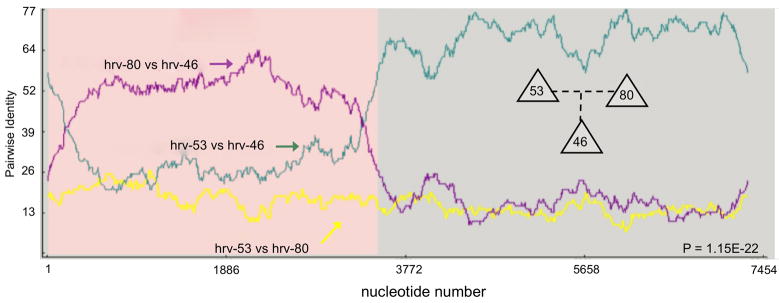
Human rhinovirus (HRV) infection is the cause of about one half of asthma and chronic obstructive pulmonary disease exacerbations. With more than 100 serotypes in the HRV reference set, an effort was undertaken to sequence their complete genomes so as to understand the diversity, structural variation, and evolution of the virus. Analysis revealed conserved motifs, hypervariable regions, a potential fourth HRV species, within-serotype variation in field isolates, a nonscanning internal ribosome entry site, and evidence for HRV recombination. Techniques have now been developed using next-generation sequencing to generate complete genomes from patient isolates with high throughput, deep coverage, and low costs. Thus relationships can now be sought between obstructive lung phenotypes and variation in HRV genomes in infected patients and potential novel therapeutic strategies developed based on HRV sequence.
Copyright (c) 2010 American Academy of Allergy, Asthma & Immunology. Published by Mosby, Inc. All rights reserved.
Figures
References
-
- Gwaltney JM, Jr, Hendley JO, Simon G, Jordan WS., Jr Rhinovirus infections in an industrial population. I. The occurrence of illness. N Engl J Med. 1966;275:1261–8. - PubMed
-
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Campbell MJ, Josephs LK, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med. 1996;154:654–60. - PubMed
-
- Varkey JB, Varkey B. Viral infections in patients with chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2008;14:89–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
