

Stat3: linking inflammation to epithelial cancer - more than a "gut" feeling?
- PMID: 20478049
- PMCID: PMC2887830
- DOI: 10.1186/1747-1028-5-14
Stat3: linking inflammation to epithelial cancer - more than a "gut" feeling?
Abstract
Inflammation is an important environmental factor that promotes tumourigenesis and the progression of established cancerous lesions, and recent studies have started to dissect the mechanisms linking the two pathologies. These inflammatory and infectious conditions trigger immune and stromal cell release of soluble mediators which facilitate survival and proliferation of tumour cells in a paracrine manner. In addition, (epi-)genetic mutations affecting oncogenes, tumour-suppressor genes, chromosomal rearrangements and amplifications trigger the release of inflammatory mediators within the tumour microenvironment to promote neoplastic growth in an autocrine manner. These two pathways converge in tumour cells and result in activation of the latent signal transducer and activator of transcription 3 (Stat3) which mediates a transcriptional response favouring survival, proliferation and angiogenesis. The abundance of cytokines that activate Stat3 within the tumour microenvironment, which comprises of members of the interleukin (IL) IL6, IL10 and IL17/23 families, underpins a signaling network that simultaneously promotes the growth of neoplastic epithelium, fuels inflammation and suppresses the host's anti-tumour immune response. Accordingly, aberrant and persistent Stat3 activation is a frequent observation in human cancers of epithelial origin and is often associated with poor outcome.Here we summarize insights gained from mice harbouring mutations in components of the Stat3 signaling cascade and in particular of gp130, the shared receptor for the IL6 family of cytokines. We focus on the various feed-back and feed-forward loops in which Stat3 provides the signaling node in cells of the tumour and its microenvironment thereby functionally linking excessive inflammation to neoplastic growth. Although these observations are particularly pertinent to gastrointestinal tumours, we suggest that the tumour's addiction to persistent Stat3 activation is likely to also impact on other epithelial cell-derived cancers. These insights provide clues to the judicious interference of the gp130/Stat3 signaling cascade in therapeutically targeting cancer.
Figures
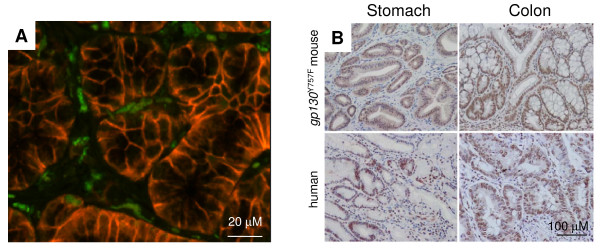
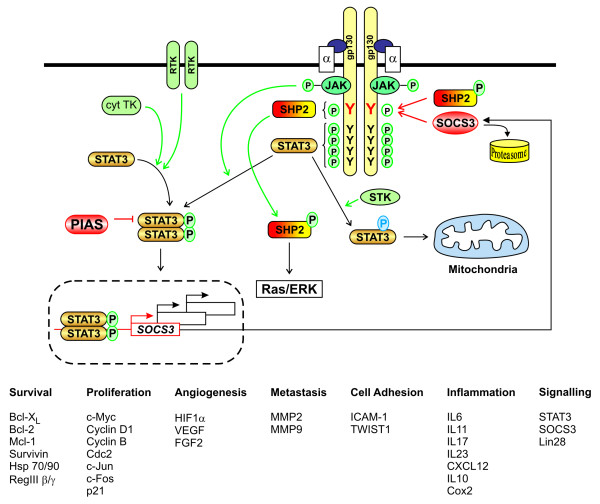
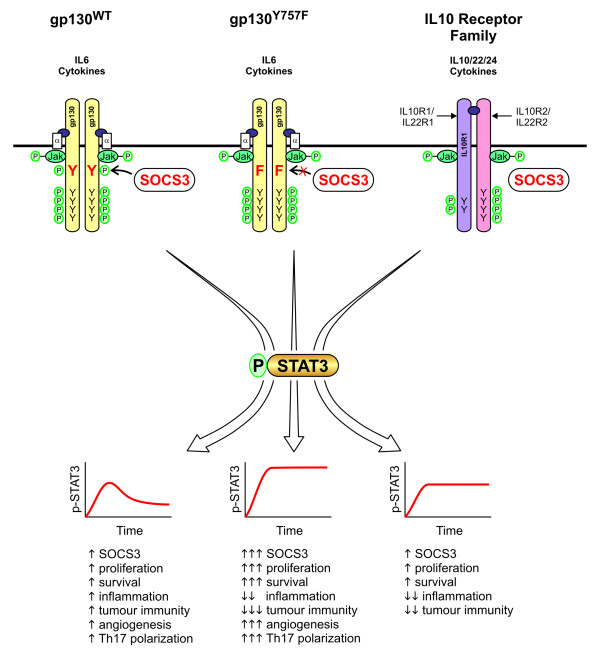
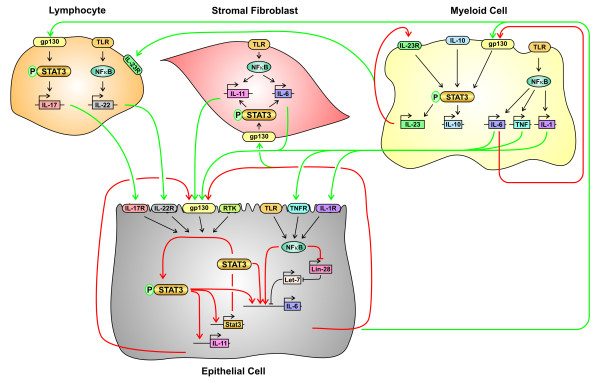
References
-
- zur Hausen H. Papovaviruses and human tumors. Haematol Blood Transfus. 1983;28:289–292. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
