

An integrative multi-dimensional genetic and epigenetic strategy to identify aberrant genes and pathways in cancer
- PMID: 20478067
- PMCID: PMC2880289
- DOI: 10.1186/1752-0509-4-67
An integrative multi-dimensional genetic and epigenetic strategy to identify aberrant genes and pathways in cancer
Abstract
Background: Genomics has substantially changed our approach to cancer research. Gene expression profiling, for example, has been utilized to delineate subtypes of cancer, and facilitated derivation of predictive and prognostic signatures. The emergence of technologies for the high resolution and genome-wide description of genetic and epigenetic features has enabled the identification of a multitude of causal DNA events in tumors. This has afforded the potential for large scale integration of genome and transcriptome data generated from a variety of technology platforms to acquire a better understanding of cancer.
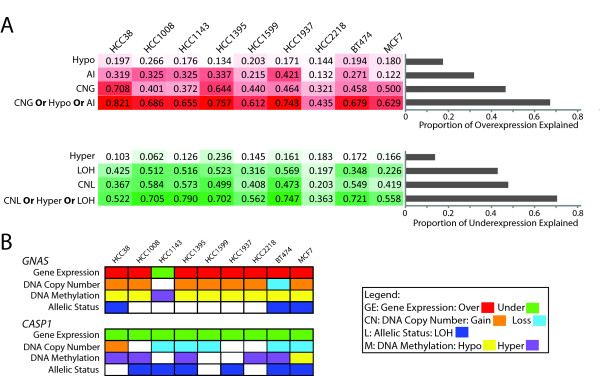
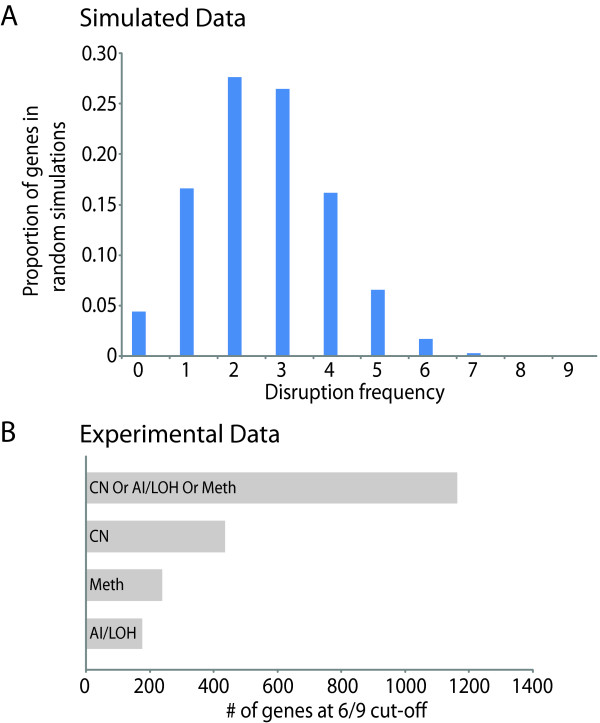
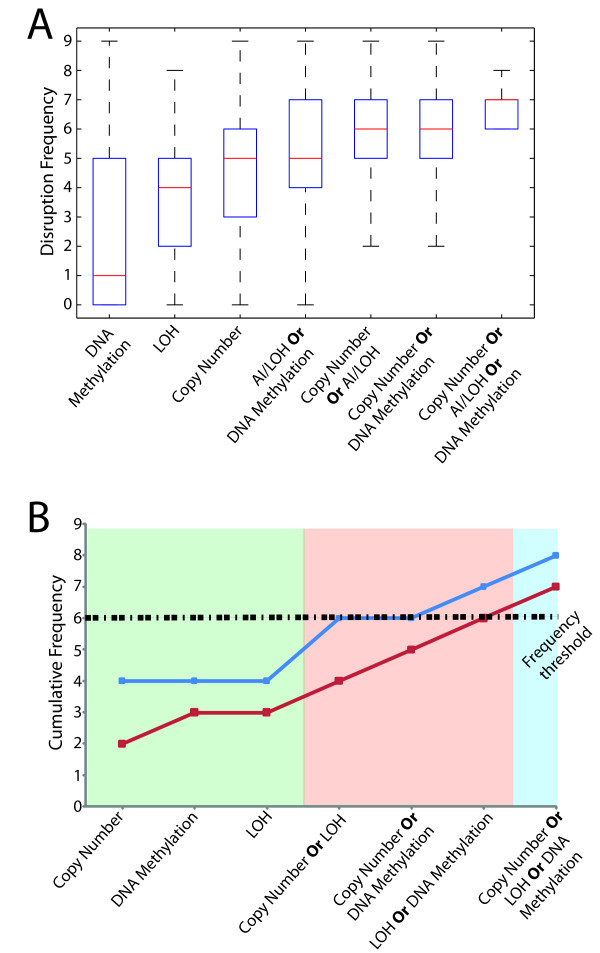
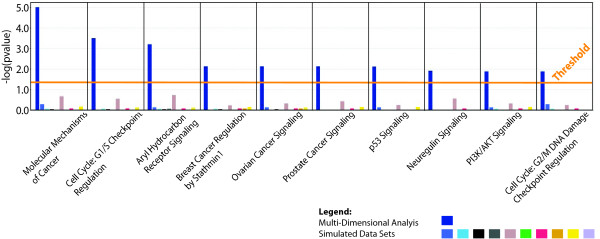
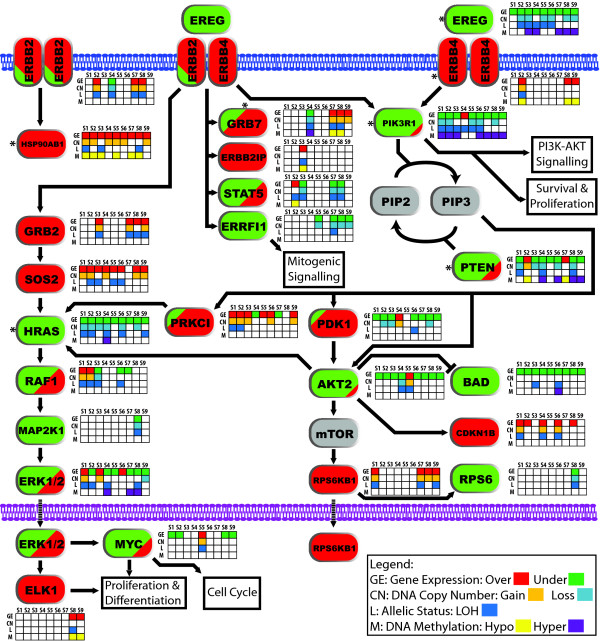
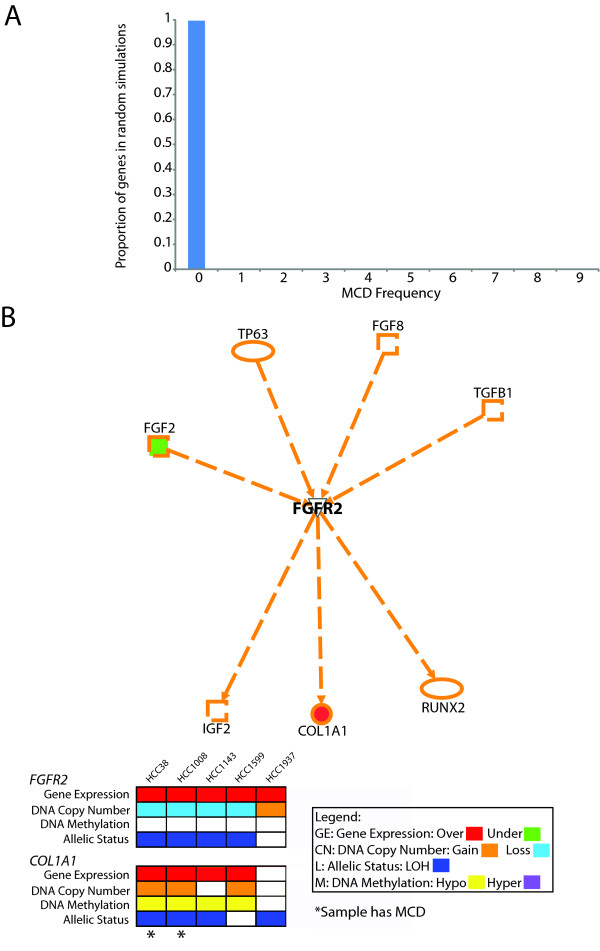
Results: Here we show how multi-dimensional genomics data analysis would enable the deciphering of mechanisms that disrupt regulatory/signaling cascades and downstream effects. Since not all gene expression changes observed in a tumor are causal to cancer development, we demonstrate an approach based on multiple concerted disruption (MCD) analysis of genes that facilitates the rational deduction of aberrant genes and pathways, which otherwise would be overlooked in single genomic dimension investigations.
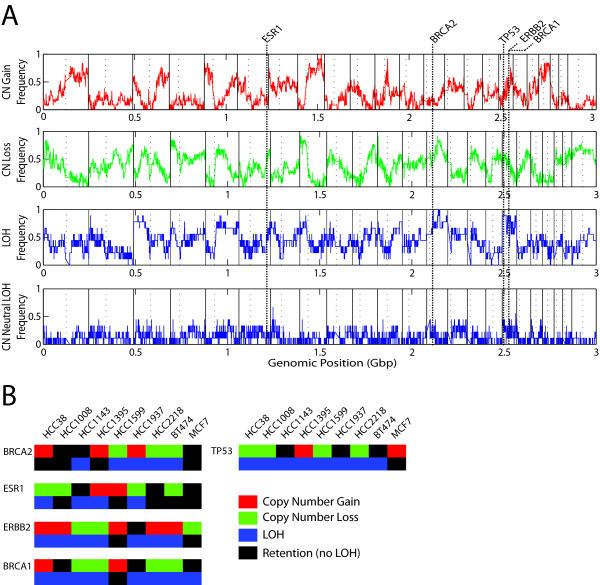
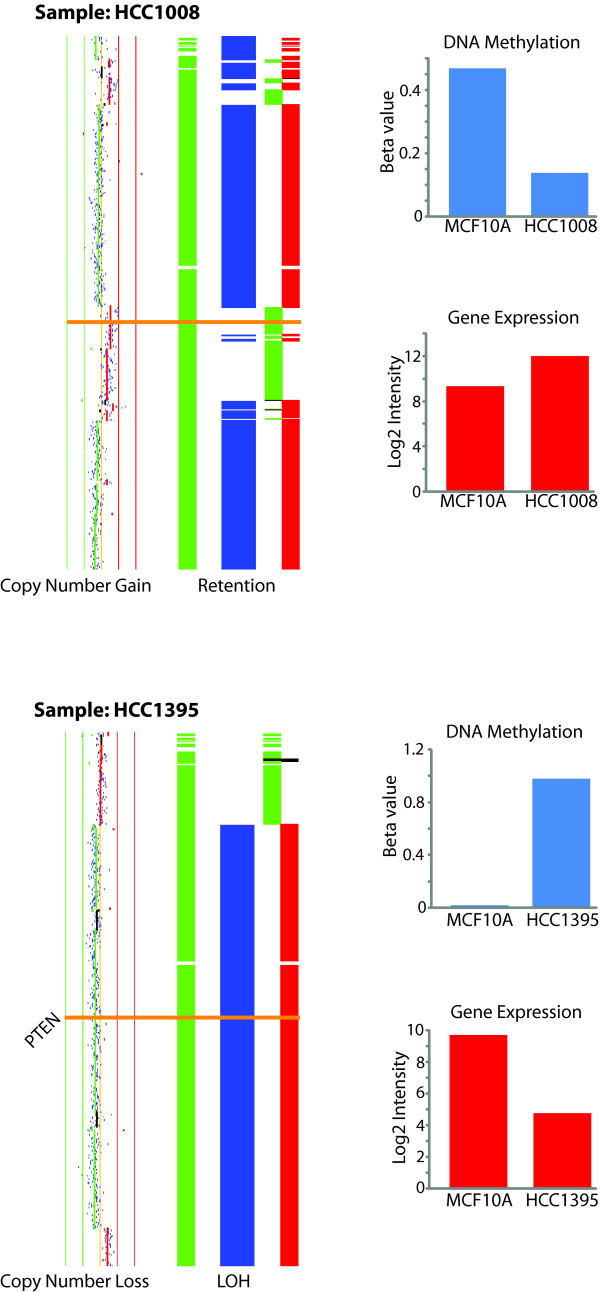
Conclusions: Notably, this is the first comprehensive study of breast cancer cells by parallel integrative genome wide analyses of DNA copy number, LOH, and DNA methylation status to interpret changes in gene expression pattern. Our findings demonstrate the power of a multi-dimensional approach to elucidate events which would escape conventional single dimensional analysis and as such, reduce the cohort sample size for cancer gene discovery.
Figures
References
-
- Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362(9381):362–369. doi: 10.1016/S0140-6736(03)14023-8. - DOI - PubMed
-
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, Rijn M van de, Jeffrey SS. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
