

A malignant hyperthermia-inducing mutation in RYR1 (R163C): consequent alterations in the functional properties of DHPR channels
- PMID: 20479108
- PMCID: PMC2888063
- DOI: 10.1085/jgp.200910329
A malignant hyperthermia-inducing mutation in RYR1 (R163C): consequent alterations in the functional properties of DHPR channels
Abstract
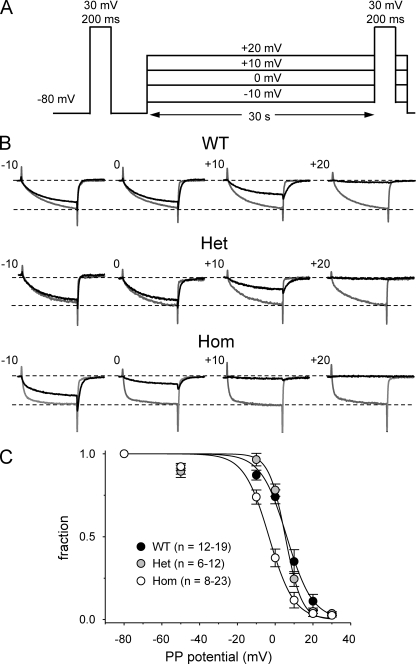
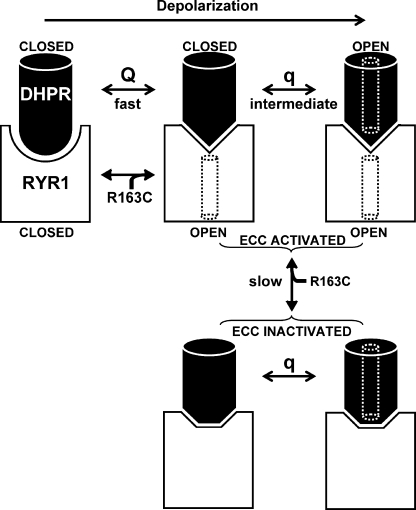
Bidirectional communication between the 1,4-dihydropyridine receptor (DHPR) in the plasma membrane and the type 1 ryanodine receptor (RYR1) in the sarcoplasmic reticulum (SR) is responsible for both skeletal-type excitation-contraction coupling (voltage-gated Ca(2+) release from the SR) and increased amplitude of L-type Ca(2+) current via the DHPR. Because the DHPR and RYR1 are functionally coupled, mutations in RYR1 that are linked to malignant hyperthermia (MH) may affect DHPR activity. For this reason, we investigated whether cultured myotubes originating from mice carrying an MH-linked mutation in RYR1 (R163C) had altered voltage-gated Ca(2+) release from the SR, membrane-bound charge movement, and/or L-type Ca(2+) current. In myotubes homozygous (Hom) for the R163C mutation, voltage-gated Ca(2+) release from the SR was substantially reduced and shifted ( approximately 10 mV) to more hyperpolarizing potentials compared with wild-type (WT) myotubes. Intramembrane charge movements of both Hom and heterozygous (Het) myotubes displayed hyperpolarizing shifts similar to that observed in voltage-gated SR Ca(2+) release. The current-voltage relationships for L-type currents in both Hom and Het myotubes were also shifted to more hyperpolarizing potentials ( approximately 7 and 5 mV, respectively). Compared with WT myotubes, Het and Hom myotubes both displayed a greater sensitivity to the L-type channel agonist +/-Bay K 8644 (10 microM). In general, L-type currents in WT, Het, and Hom myotubes inactivated modestly after 30-s prepulses to -50, -10, 0, 10, 20, and 30 mV. However, L-type currents in Hom myotubes displayed a hyperpolarizing shift in inactivation relative to L-type currents in either WT or Het myotubes. Our present results indicate that mutations in RYR1 can alter DHPR activity and raise the possibility that this altered DHPR function may contribute to MH episodes.
Figures
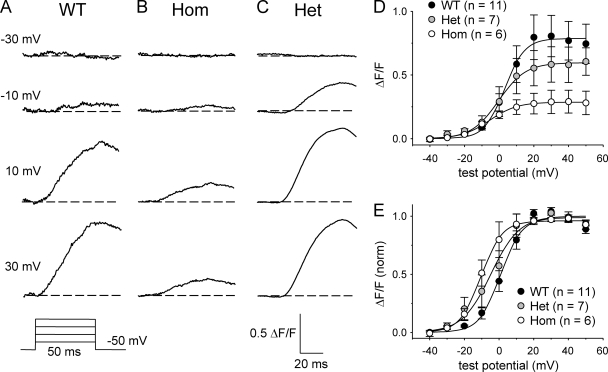
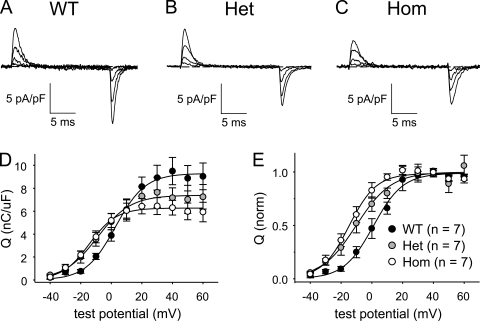
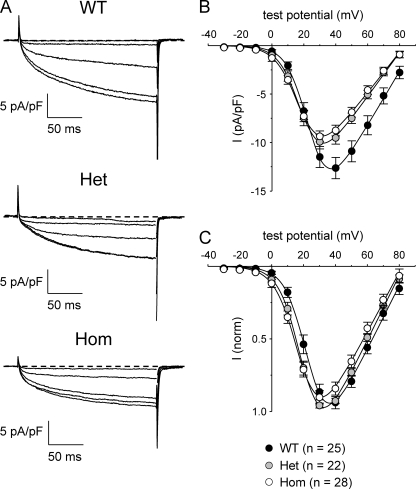
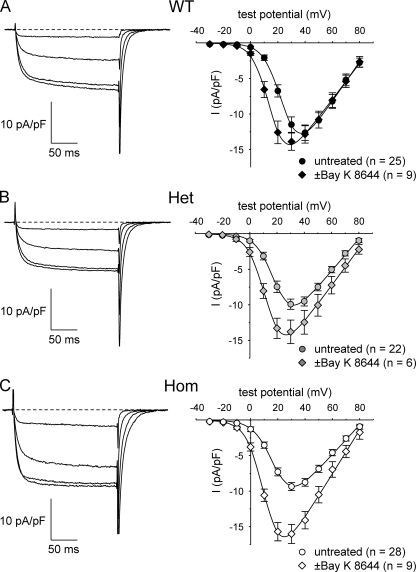
Similar articles
-
A malignant hyperthermia-inducing mutation in RYR1 (R163C): alterations in Ca2+ entry, release, and retrograde signaling to the DHPR.J Gen Physiol. 2010 Jun;135(6):619-28. doi: 10.1085/jgp.200910328. Epub 2010 May 17. J Gen Physiol. 2010. PMID: 20479110 Free PMC article.
-
Ryanodine modification of RyR1 retrogradely affects L-type Ca(2+) channel gating in skeletal muscle.J Muscle Res Cell Motil. 2009;30(5-6):217-23. doi: 10.1007/s10974-009-9190-0. Epub 2009 Oct 3. J Muscle Res Cell Motil. 2009. PMID: 19802526 Free PMC article.
-
Functional analysis of the R1086H malignant hyperthermia mutation in the DHPR reveals an unexpected influence of the III-IV loop on skeletal muscle EC coupling.Am J Physiol Cell Physiol. 2004 Oct;287(4):C1094-102. doi: 10.1152/ajpcell.00173.2004. Epub 2004 Jun 16. Am J Physiol Cell Physiol. 2004. PMID: 15201141
-
Malignant hyperthermia and excitation-contraction coupling.Acta Physiol Scand. 2001 Mar;171(3):367-78. doi: 10.1046/j.1365-201x.2001.00840.x. Acta Physiol Scand. 2001. PMID: 11412150 Review.
-
Bridging the myoplasmic gap II: more recent advances in skeletal muscle excitation-contraction coupling.J Exp Biol. 2016 Jan;219(Pt 2):175-82. doi: 10.1242/jeb.124123. J Exp Biol. 2016. PMID: 26792328 Review.
Cited by
-
Structural mechanism of two gain-of-function cardiac and skeletal RyR mutations at an equivalent site by cryo-EM.Sci Adv. 2020 Jul 29;6(31):eabb2964. doi: 10.1126/sciadv.abb2964. eCollection 2020 Jul. Sci Adv. 2020. PMID: 32832689 Free PMC article.
-
Functional and biochemical properties of ryanodine receptor type 1 channels from heterozygous R163C malignant hyperthermia-susceptible mice.Mol Pharmacol. 2011 Mar;79(3):420-31. doi: 10.1124/mol.110.067959. Epub 2010 Dec 14. Mol Pharmacol. 2011. PMID: 21156754 Free PMC article.
-
Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: a comprehensive scoping review of works published 1990-2019.Orphanet J Rare Dis. 2020 May 7;15(1):113. doi: 10.1186/s13023-020-01384-x. Orphanet J Rare Dis. 2020. PMID: 32381029 Free PMC article.
-
Ca(V)1.1: The atypical prototypical voltage-gated Ca²⁺ channel.Biochim Biophys Acta. 2013 Jul;1828(7):1587-97. doi: 10.1016/j.bbamem.2012.09.007. Epub 2012 Sep 13. Biochim Biophys Acta. 2013. PMID: 22982493 Free PMC article. Review.
-
A malignant hyperthermia-inducing mutation in RYR1 (R163C): alterations in Ca2+ entry, release, and retrograde signaling to the DHPR.J Gen Physiol. 2010 Jun;135(6):619-28. doi: 10.1085/jgp.200910328. Epub 2010 May 17. J Gen Physiol. 2010. PMID: 20479110 Free PMC article.
References
-
- Adnet P.J., Krivosic-Horber R.M., Adamantidis M.M., Haudecoeur G., Reyford H., Imbenotte M. 1992. Effect of Bay K 8644 on the magnitude of isoflurane and halothane contracture of skeletal muscle from patients susceptible to malignant hyperthermia. Anesthesiology. 76:544–549 10.1097/00000542-199204000-00010 - DOI - PubMed
-
- Ahern C.A., Sheridan D.C., Cheng W., Mortenson L., Nataraj P., Allen P.D., De Waard M., Coronado R. 2003. Ca2+ current and charge movements in skeletal myotubes promoted by the β-subunit of the dihydropyridine receptor in the absence of ryanodine receptor type 1. Biophys. J. 84:942–959 10.1016/S0006-3495(03)74911-X - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
