

Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease
- PMID: 20481466
- PMCID: PMC2885827
- DOI: 10.1021/bi1004798
Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease
Abstract
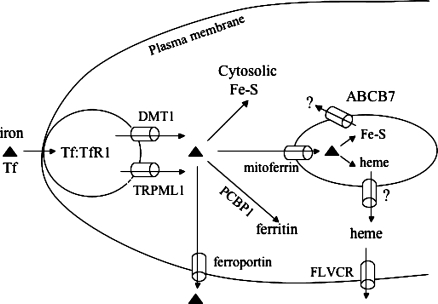
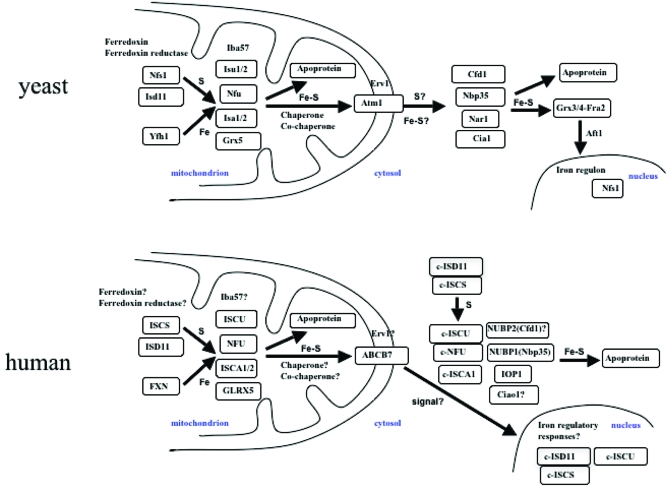
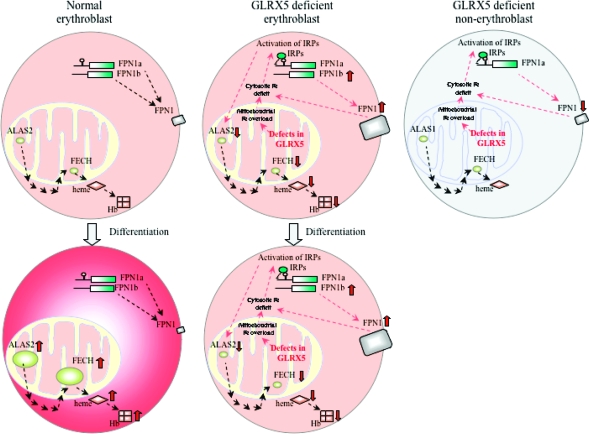
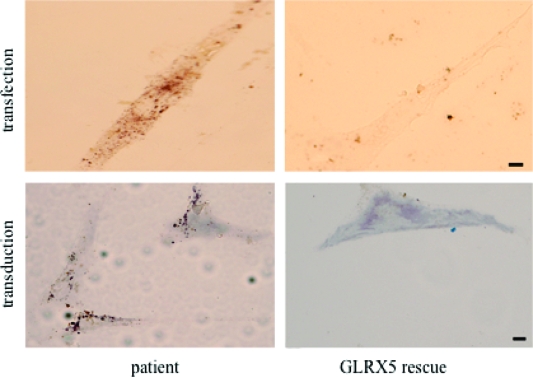
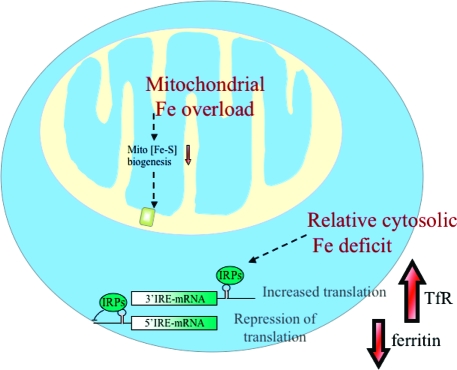
Iron-sulfur (Fe-S) proteins contain prosthetic groups consisting of two or more iron atoms bridged by sulfur ligands, which facilitate multiple functions, including redox activity, enzymatic function, and maintenance of structural integrity. More than 20 proteins are involved in the biosynthesis of iron-sulfur clusters in eukaryotes. Defective Fe-S cluster synthesis not only affects activities of many iron-sulfur enzymes, such as aconitase and succinate dehydrogenase, but also alters the regulation of cellular iron homeostasis, causing both mitochondrial iron overload and cytosolic iron deficiency. In this work, we review human Fe-S cluster biogenesis and human diseases that are caused by defective Fe-S cluster biogenesis. Fe-S cluster biogenesis takes place essentially in every tissue of humans, and products of human disease genes, including frataxin, GLRX5, ISCU, and ABCB7, have important roles in the process. However, the human diseases, Friedreich ataxia, glutaredoxin 5-deficient sideroblastic anemia, ISCU myopathy, and ABCB7 sideroblastic anemia/ataxia syndrome, affect specific tissues, while sparing others. Here we discuss the phenotypes caused by mutations in these different disease genes, and we compare the underlying pathophysiology and discuss the possible explanations for tissue-specific pathology in these diseases caused by defective Fe-S cluster biogenesis.
Figures
References
-
- Yin L.; Wu N.; Curtin J. C.; Qatanani M.; Szwergold N. R.; Reid R. A.; Waitt G. M.; Parks D. J.; Pearce K. H.; Wisely G. B.; Lazar M. A. (2007) Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789. - PubMed
-
- Faller M.; Matsunaga M.; Yin S.; Loo J. A.; Guo F. (2007) Heme is involved in microRNA processing. Nat. Struct. Mol. Biol. 14, 23–29. - PubMed
-
- Hentze M. W.; Muckenthaler M. U.; Andrews N. C. (2004) Balancing acts: Molecular control of mammalian iron metabolism. Cell 117, 285–297. - PubMed
-
- De Domenico I.; McVey Ward D.; Kaplan J. (2008) Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 9, 72–81. - PubMed
-
- Ganz T. (2008) Iron homeostasis: Fitting the puzzle pieces together. Cell Metab. 7, 288–290. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
