

Towards a comprehensive structural variation map of an individual human genome
- PMID: 20482838
- PMCID: PMC2898065
- DOI: 10.1186/gb-2010-11-5-r52
Towards a comprehensive structural variation map of an individual human genome
Abstract
Background: Several genomes have now been sequenced, with millions of genetic variants annotated. While significant progress has been made in mapping single nucleotide polymorphisms (SNPs) and small (<10 bp) insertion/deletions (indels), the annotation of larger structural variants has been less comprehensive. It is still unclear to what extent a typical genome differs from the reference assembly, and the analysis of the genomes sequenced to date have shown varying results for copy number variation (CNV) and inversions.
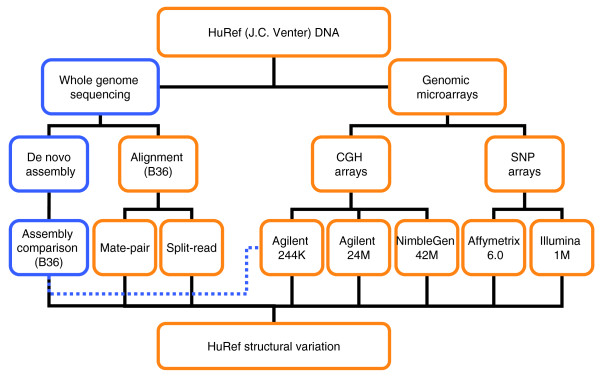
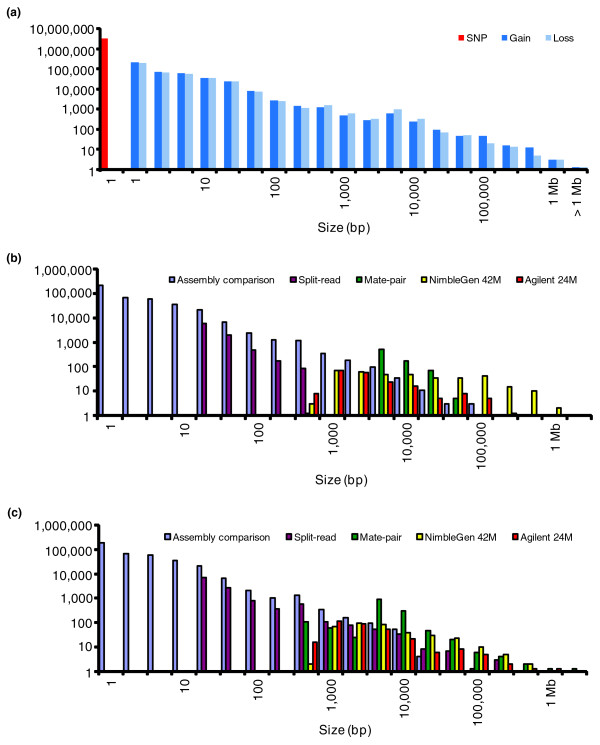
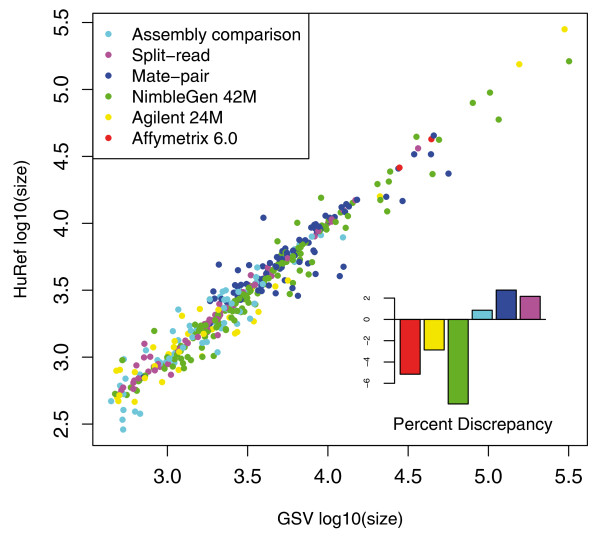
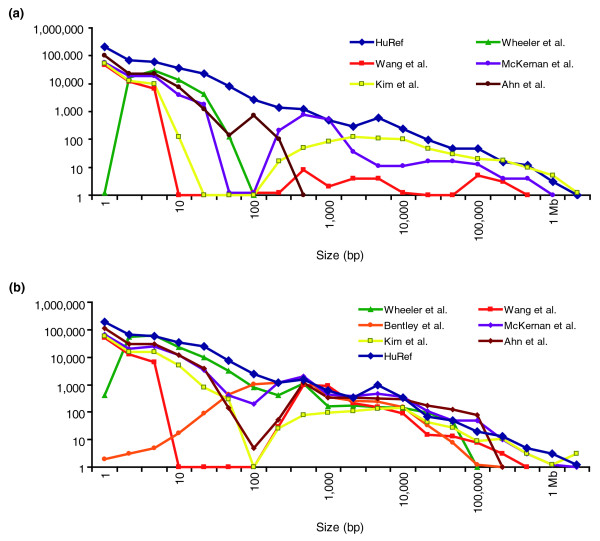
Results: We have combined computational re-analysis of existing whole genome sequence data with novel microarray-based analysis, and detect 12,178 structural variants covering 40.6 Mb that were not reported in the initial sequencing of the first published personal genome. We estimate a total non-SNP variation content of 48.8 Mb in a single genome. Our results indicate that this genome differs from the consensus reference sequence by approximately 1.2% when considering indels/CNVs, 0.1% by SNPs and approximately 0.3% by inversions. The structural variants impact 4,867 genes, and >24% of structural variants would not be imputed by SNP-association.
Conclusions: Our results indicate that a large number of structural variants have been unreported in the individual genomes published to date. This significant extent and complexity of structural variants, as well as the growing recognition of their medical relevance, necessitate they be actively studied in health-related analyses of personal genomes. The new catalogue of structural variants generated for this genome provides a crucial resource for future comparison studies.
Figures
References
-
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL. et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. - DOI - PMC - PubMed
-
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. - DOI - PubMed
-
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. - DOI - PMC - PubMed
-
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y, Feng B, Li H, Lu Y, Fang X, Liang H, Du Z, Li D, Zhao Y, Hu Y, Yang Z, Zheng H, Hellmann I, Inouye M, Pool J, Yi X, Zhao J, Duan J, Zhou Y, Qin J. et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. - DOI - PMC - PubMed
-
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, Cook L, Abbott R, Larson DE, Koboldt DC, Pohl C, Smith S, Hawkins A, Abbott S, Locke D, Hillier LW, Miner T, Fulton L, Magrini V, Wylie T, Glasscock J, Conyers J, Sander N, Shi X, Osborne JR, Minx P. et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
