

Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl
- PMID: 20482842
- PMCID: PMC2893099
- DOI: 10.1186/1476-4598-9-112
Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl
Erratum in
-
Correction to: Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-κB signaling and depleting Bcr-Abl.Mol Cancer. 2021 Dec 31;20(1):172. doi: 10.1186/s12943-021-01414-7. Mol Cancer. 2021. PMID: 34969396 Free PMC article. No abstract available.
Abstract
Background: Chronic myelogenous leukemia (CML) is characterized by the chimeric tyrosine kinase Bcr-Abl. Bcr-Abl-T315I is the notorious point mutation that causes resistance to imatinib and the second generation tyrosine kinase inhibitors, leading to poor prognosis. CML blasts have constitutive p65 (RelA NF-kappaB) transcriptional activity, and NF-kappaB may be a potential target for molecular therapies in CML that may also be effective against CML cells with Bcr-Abl-T315I.
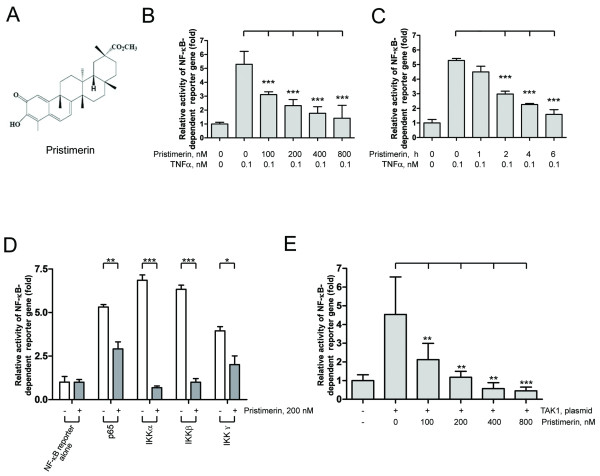
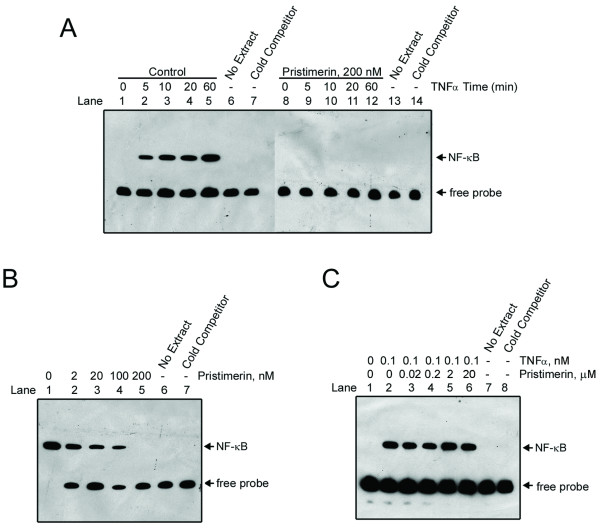
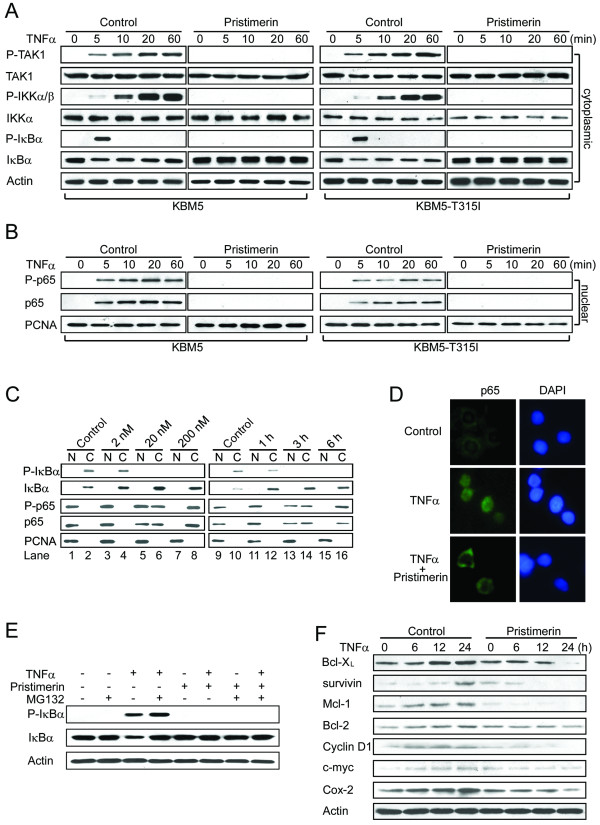
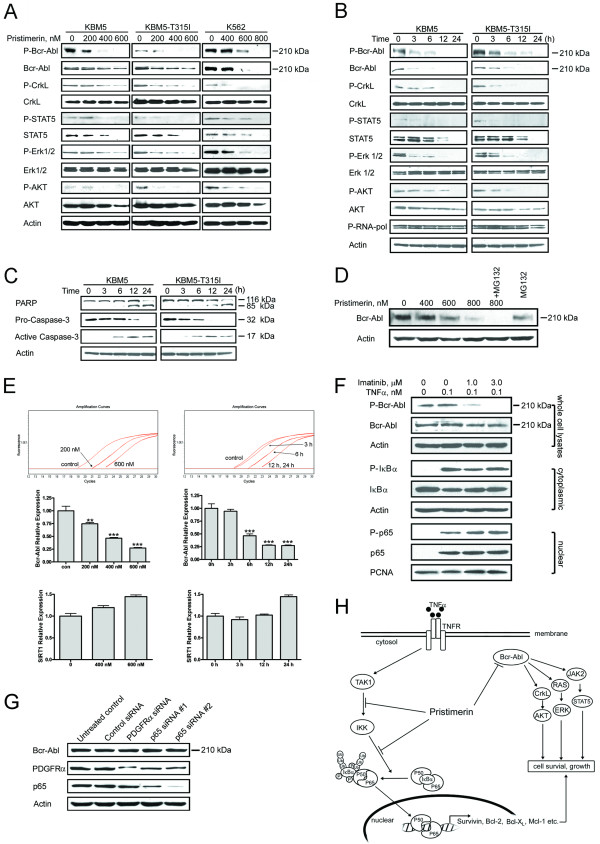
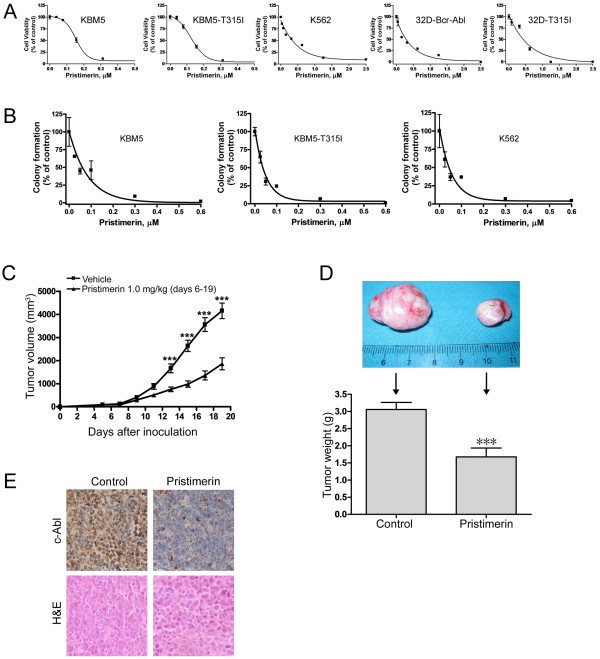
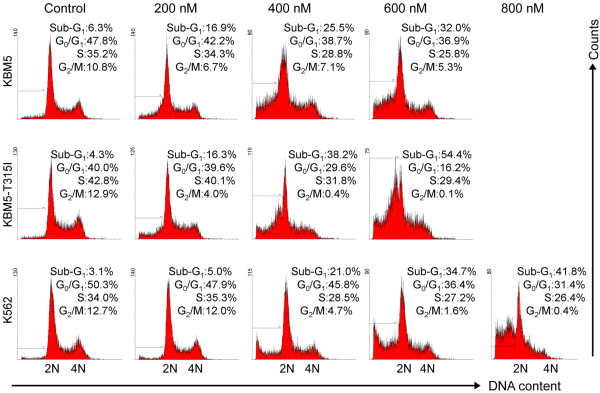
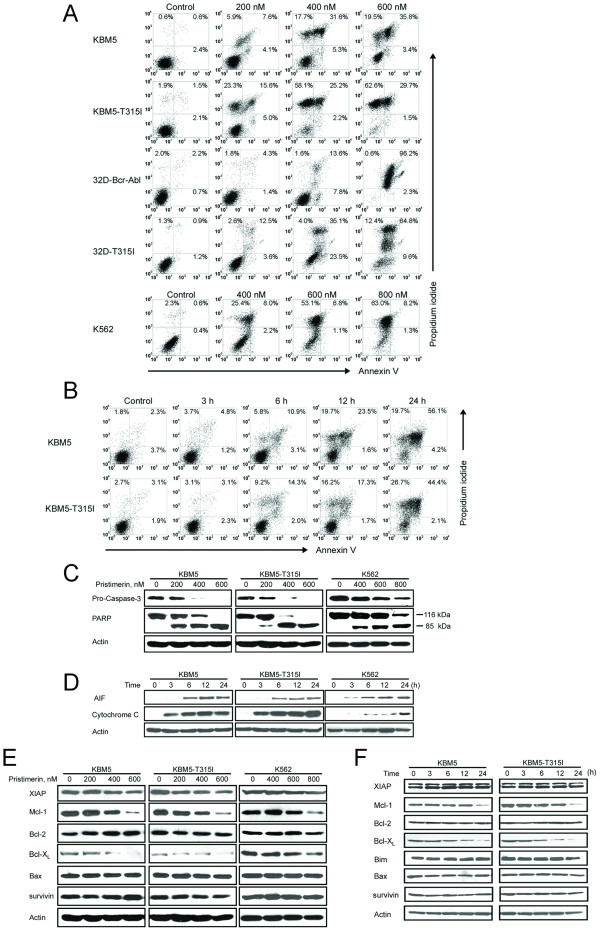
Results: In this report, we discovered that pristimerin, a quinonemethide triterpenoid isolated from Celastraceae and Hippocrateaceae, inhibited growth and induced apoptosis in CML cells, including the cells harboring Bcr-Abl-T315I mutation. Additionally, pristimerin inhibited the growth of imatinib-resistant Bcr-Abl-T315I xenografts in nude mice. Pristimerin blocked the TNFalpha-induced IkappaBalpha phosphorylation, translocation of p65, and expression of NF-kappaB-regulated genes. Pristimerin inhibited two steps in NF-kappaB signaling: TAK1TauIKK and IKKTauIkappaBalpha. Pristimerin potently inhibited two pairs of CML cell lines (KBM5 versus KBM5-T315I, 32D-Bcr-Abl versus 32D-Bcr-Abl-T315I) and primary cells from a CML patient with acquired resistance to imatinib. The mRNA and protein levels of Bcr-Abl in imatinib-sensitive (KBM5) or imatinib-resistant (KBM5-T315I) CML cells were reduced after pristimerin treatment. Further, inactivation of Bcr-Abl by imatinib pretreatment did not abrogate the TNFalpha-induced NF-kappaB activation while silencing p65 by siRNA did not affect the levels of Bcr-Abl, both results together indicating that NF-kappaB inactivation and Bcr-Abl inhibition may be parallel independent pathways.
Conclusion: To our knowledge, this is the first report to show that pristimerin is effective in vitro and in vivo against CML cells, including those with the T315I mutation. The mechanisms may involve inhibition of NF-kappaB and Bcr-Abl. We concluded that pristimerin could be a lead compound for further drug development to overcome imatinib resistance in CML patients.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
