

Protein thermostability calculations using alchemical free energy simulations
- PMID: 20483340
- PMCID: PMC2872215
- DOI: 10.1016/j.bpj.2010.01.051
Protein thermostability calculations using alchemical free energy simulations
Abstract
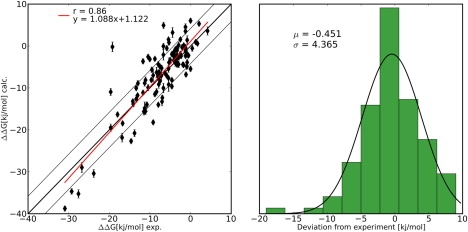
Thermal stability of proteins is crucial for both biotechnological and therapeutic applications. Rational protein engineering therefore frequently aims at increasing thermal stability by introducing stabilizing mutations. The accurate prediction of the thermodynamic consequences caused by mutations, however, is highly challenging as thermal stability changes are caused by alterations in the free energy of folding. Growing computational power, however, increasingly allows us to use alchemical free energy simulations, such as free energy perturbation or thermodynamic integration, to calculate free energy differences with relatively high accuracy. In this article, we present an automated protocol for setting up alchemical free energy calculations for mutations of naturally occurring amino acids (except for proline) that allows an unprecedented, automated screening of large mutant libraries. To validate the developed protocol, we calculated thermodynamic stability differences for 109 mutations in the microbial Ribonuclease Barnase. The obtained quantitative agreement with experimental data illustrates the potential of the approach in protein engineering and design.
Copyright 2010 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
