

Review
doi: 10.1038/nrn2841.
22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia
Affiliations
- PMID: 20485365
- PMCID: PMC2977984
- DOI: 10.1038/nrn2841
Item in Clipboard
Review
22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia
Nat Rev Neurosci.
2010 Jun.
Abstract
Recent studies are beginning to paint a clear and consistent picture of the impairments in psychological and cognitive competencies that are associated with microdeletions in chromosome 22q11.2. These studies have highlighted a strong link between this genetic lesion and schizophrenia. Parallel studies in humans and animal models are starting to uncover the complex genetic and neural substrates altered by the microdeletion. In addition to offering a deeper understanding of the effects of this genetic lesion, these findings may guide analysis of other copy-number variants associated with cognitive dysfunction and psychiatric disorders.
Figures
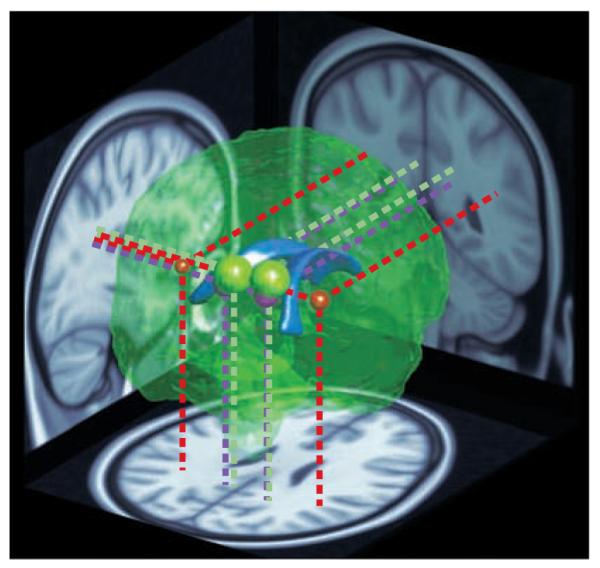
A reconstruction of the brain viewed from the posterior right side. The lateral ventricles (shown in blue) are depicted in the centre as a point of reference. The coloured spheres indicate the location and approximate extent of major clusters of reduced fractional anisotropy (FA) — a measure of neural connectivity — in children with 22q11.2 deletion syndrome (22q11.2DS), as reported by REF. (red), REF. (green) and REF. (purple). The data were obtained using diffusion tensor imaging, an MRI technique that measures the directionality of water diffusion in the brain as an indicator of the organization and integrity of neuronal tracts wrapped in myelin (white matter). The location and size of the coloured spheres depict the sites of peak FA differences and their extent. Projections onto brain slices (dotted lines) indicate the positions of the clusters in major white matter tracts. The red spheres represent the left and right extremities in a wedge-shaped cluster reported by REF. . The entire area between them showed higher FA in children with 22q11.2DS compared with typical developing controls, but because this large cluster would completely envelope the other regions, it is not represented here in its entirety. In summary, three separate studies from two different teams have reported similar, and in one case directly overlapping, differences in neural connectivity in children and young adults with 22q11.2DS.
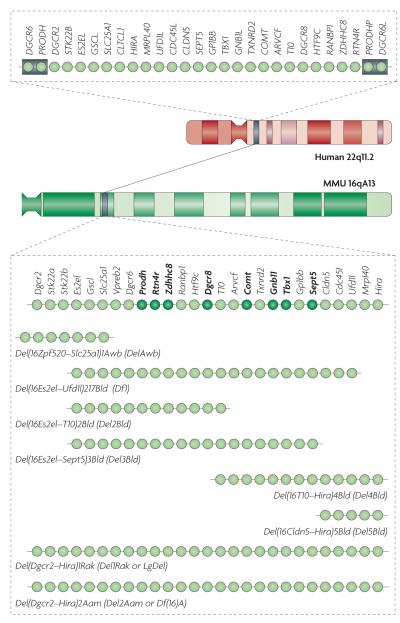
The chromosomal location and genetic organization of the 22q11.2 locus in humans is shown at the top. Each light green circle represents one gene. This 1.5 megabase (Mb) region is flanked by low-copy-repeat sequences (represented by grey boxes), making it prone to non-homologous recombination. The syntenic region of mouse chromosome 16 (MMU 16qA13) and the genetic organization of the corresponding orthologues are shown. Single-gene deletion models that have been characterized as heterozygotes for neuronal and behavioural abnormalities are indicated by dark green circles. Also shown are the various multigene deletion models that have been characterized for neuronal and behavioural abnormalities labelled using their Mouse Genome Database (MGD) allele symbols and commonly used synonyms. They include mice deficient for seven genes spanning ~150 kilobases (kb) of the 1.5 Mb deletion syntenic region (Del(16Zpf520–Slc25a1)1Awb mice (also known as DelAwb mice)); a 22q11.2 model spanning ~1 Mb and containing 18 orthologues of the human genes in the 1.5 Mb deletion (Del(16Es2el–Ufd1l)217Bld mice (also known as Df1 mice)),; mice with a hemizygous deletion spanning ~1.3 Mb and containing all but one of the orthologous genes in the 1.5 Mb deletion (Del(Dgcr2–Hira)1Rak mice (also known as Del1Rak or LgDel mice)) and mice with a hemizygous deletion syntenic to the human 1.5 Mb deletion (Del(Dgcr2–Hira)2Aam mice). ARVCF, armadillo repeat gene deleted in velocardiofacial syndrome; CDC45L, cell division cycle 45-like; CLDN5, claudin 5; CLTCL1, clathrin, heavy chain-like 1; COMT, catechol-O-methyltransferase; DGCR, DiGeorge syndrome critical region; DGCR6L, DiGeorge syndrome critical region 6-like; GNB1L, guanine-nucleotide-binding protein (G protein) β-polypeptide 1-like; GP1BB, glycoprotein Ib, β-polypeptide; GSCL, goosecoid-like; HIRA, histone cell cycle regulation defective A; HTF9C, HpaII tiny fragments locus 9C (also known as TRMT2A); MRPL40, mitochondrial ribosomal protein L40; PRODHP, proline dehydrogenase pseudogene; RANBP1, RAN binding protein 1; RTN4R, reticulon 4 receptor; SEPT5, septin 5; SLC25A1, solute carrier family 25, member 1; Stk22a, serine/threonine kinase 22A (also known as Tssk1); STK22B, serine/threonine kinase 22B (also known as TSSK2); TBX1, T-box 1; TXNRD2, thioredoxin reductase 2; UFD1L, ubiquitin fusion degradation 1-like; Vpreb2, pre-B lymphocyte gene 2; ZDHHC8, zinc finger, DHHC-type containing 8.
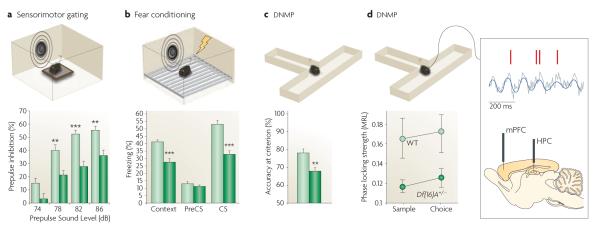
Representative results from different behavioural studies are shown from the Del(Dgcr2–Hira)2Aam mouse strain (also known as Del2Aam or Df(16)A mice) (FIG. 2). a ∣ Del(Dgcr2–Hira)2Aam mice (dark green bars) showed decreased prepulse inhibition (PPI) of the acoustic startle when compared with controls (light green bars; the asterisks represent significant differences between mutant and wild-type (WT) response). PPI is a reduction in the magnitude of the startle reflex that occurs when mice are presented with a non-startling stimulus (a prepulse) before being presented with the startling stimulus. b ∣ Del(Dgcr2–Hira)2Aam mice (dark green bars) exhibit robust deficits in both cued and contextual fear memory compared with controls (light green bars), as indicated by a decrease in the time spent exhibiting freezing behaviour. The cued and contextual fear conditioning test quantifies the ability to associate a neutral conditioned stimulus (CS; either a light or a tone) with an unconditioned stimulus (an electric shock); the cued version of the test requires the amygdala, and the context version of the test typically requires both the hippocampus (HPC) and the amygdala. c ∣ Del(Dgcr2–Hira)2Aam mice (dark green bar) are impaired in working memory-dependent cognitive performance, as shown by decreased accuracy in the delayed non-match to place (DNMP) task compared with controls (light green bar). For the DNMP task, mice are trained to enter the appropriate arm of a two-arm T-maze to obtain a food reward, the location of which varies across trials. In both the training and testing phases, short delays of various lengths are introduced, which require frontal regions of the mouse neocortex and their interaction with the HPC, for the active maintenance of information. d ∣ Del(Dgcr2–Hira)2Aam mice (dark green circles) showed reduced connectivity and synchrony in the HPC–prefrontal cortex (HPC–PFC) compared with controls (light green circles) as demonstrated by recordings from the medial PFC (mPFC) and HPC of mice while they performed a task that required working memory. An example of a field potential recording from the HPC (grey trace) showing θ-oscillations (blue trace) and spikes recorded simultaneously from a PFC neuron (red marks) is also shown. Note the robust modulation of prefrontal neuron spiking by hippocampal θ-oscillations (synchrony), which is disrupted in Del(Dgcr2–Hira)2Aam mice.
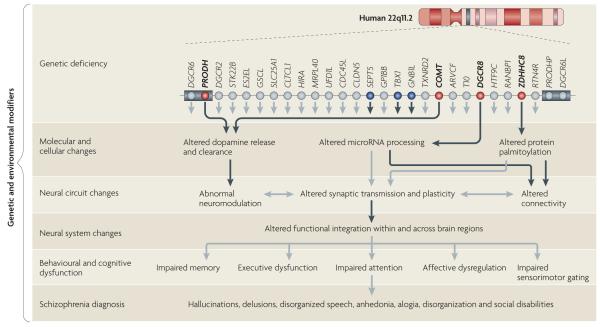
Reduced gene-dosage of more than one gene affects brain function at multiple levels in 22q11.2 deletion syndrome (22q11.2DS). For simplicity, only the 1.5 megabase (Mb) region is shown. In the upper row, red circles indicate genes (proline dehydrogenase (PRODH), catechol-O-methyltransferase (COMT), DiGeorge syndrome critical region 8 (DGCR8) and zinc finger, DHHC-type containing 8 (ZDHHC8)) that have been characterized at the molecular, cellular, neural circuit and behavioural levels. Blue circles indicate genes (septin 5 (SEPT5), T-box 1 (TBX1) and guanine-nucleotide-binding protein (G protein) β-polypeptide 1-like (GNB1L)) that were studied only at the behavioural level and were found to cause changes in heterozygous mutant mice. The lower rows show how mutations in these genes disrupt the molecular and cellular properties of neurons, and how these disruptions interact with each other across different levels and may ultimately lead to a psychiatric dysfunction. Black arrows represent experimentally determined effects of 22q11.2 genes and grey arrows represent potential links inferred from existing knowledge. The cumulative effect of the imbalance of several genes in the deletion determines the overall phenotype. Genes responsible for specific phenotypic deficits are only partially overlapping, and additional genetic and environmental modifiers may also contribute to the expression and variability of these deficits. The grey boxes represent low-copy-repeat sequences that flank the 22q11.2 locus. ARVCF, armadillo repeat gene deleted in velocardiofacial syndrome; CDC45L, cell division cycle 45-like; CLDN5, claudin 5; CLTCL1, clathrin, heavy chain-like 1; DGCR6L, DiGeorge syndrome critical region 6-like; GP1BB, glycoprotein Ib, β-polypeptide; GSCL, goosecoid-like; HIRA, histone cell cycle regulation defective A; HTF9C, HpaII tiny fragments locus 9C (also known as TRMT2A); MRPL40, mitochondrial ribosomal protein L40; PRODHP, proline dehydrogenase pseudogene; RANBP1, RAN binding protein 1; RTN4R, reticulon 4 receptor; SLC25A1, solute carrier family 25, member 1; STK22B, serine/threonine kinase 22B (also known as TSSK2); TXNRD2, thioredoxin reductase 2; UFD1L, ubiquitin fusion degradation 1-like.
References
-
- Shprintzen RJ, et al. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15:56–62. - PubMed
-
- DiGeorge A. A new concept of the cellular basis of immunity. Disabil. Rehabil. 1965;67:907–908.
-
- Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion. 22q11.2. J. Pediatr. 2005;147:90–96. - PubMed
-
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. - PubMed
-
- Botto LD, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
