

Hepatitis C virus controls interferon production through PKR activation
- PMID: 20485506
- PMCID: PMC2868028
- DOI: 10.1371/journal.pone.0010575
Hepatitis C virus controls interferon production through PKR activation
Abstract
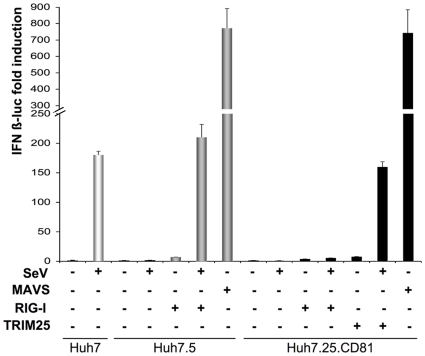
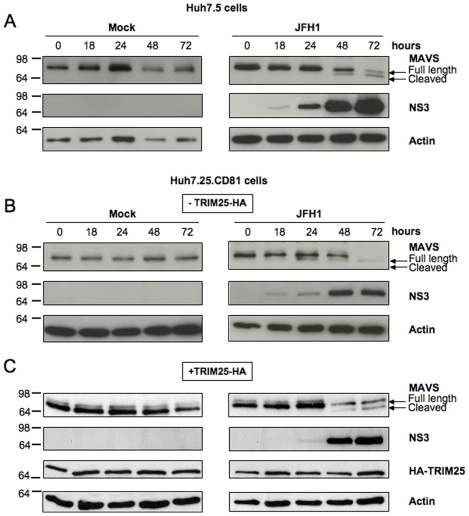
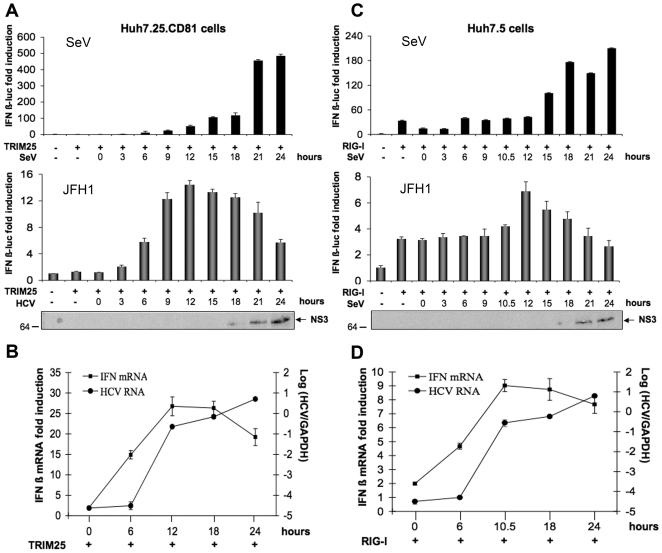
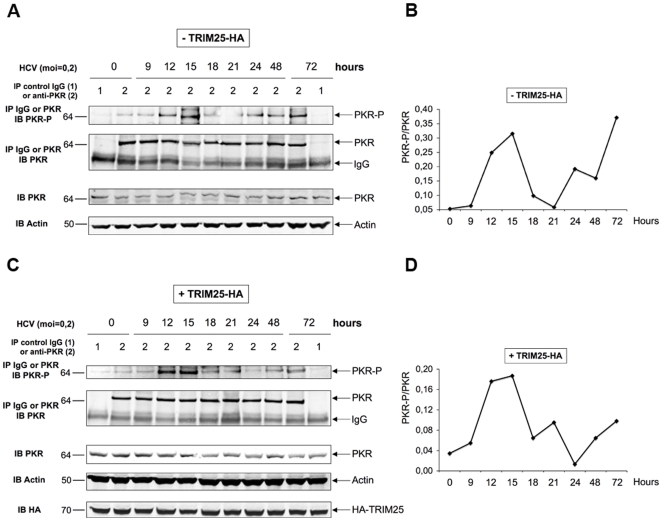
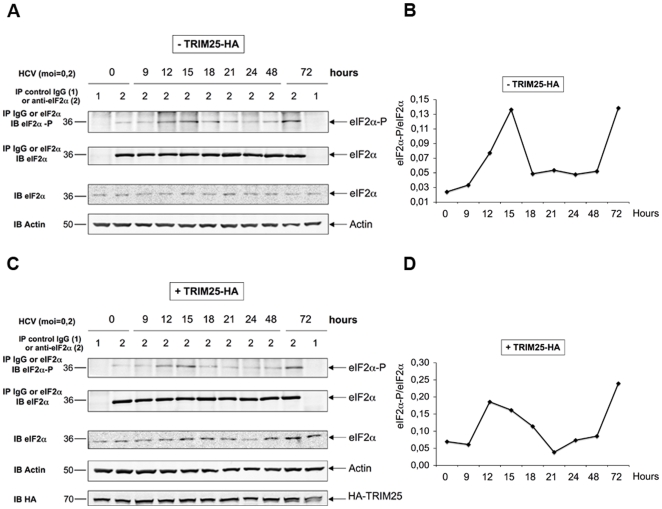
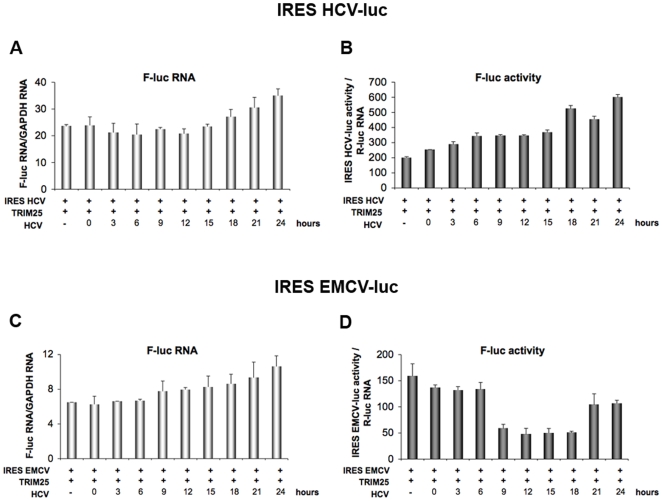
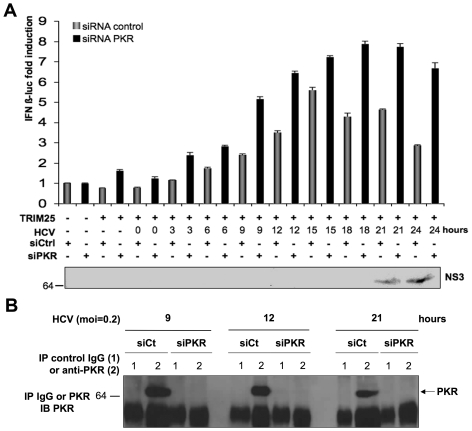
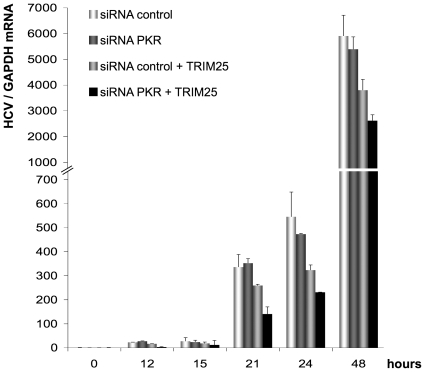
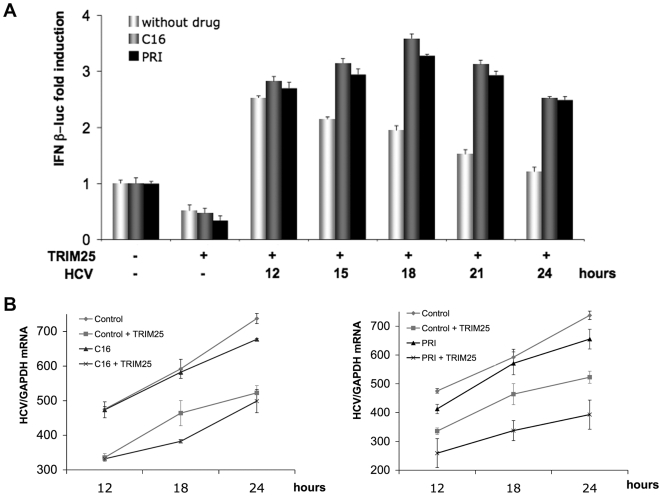
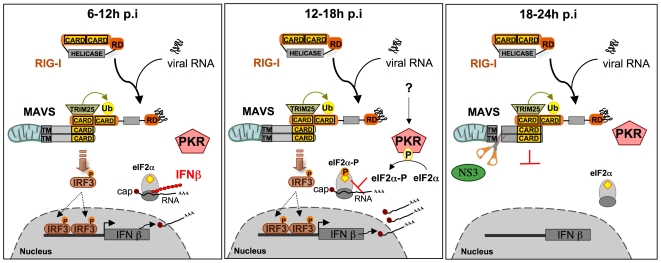
Hepatitis C virus is a poor inducer of interferon (IFN), although its structured viral RNA can bind the RNA helicase RIG-I, and activate the IFN-induction pathway. Low IFN induction has been attributed to HCV NS3/4A protease-mediated cleavage of the mitochondria-adapter MAVS. Here, we have investigated the early events of IFN induction upon HCV infection, using the cell-cultured HCV JFH1 strain and the new HCV-permissive hepatoma-derived Huh7.25.CD81 cell subclone. These cells depend on ectopic expression of the RIG-I ubiquitinating enzyme TRIM25 to induce IFN through the RIG-I/MAVS pathway. We observed induction of IFN during the first 12 hrs of HCV infection, after which a decline occurred which was more abrupt at the protein than at the RNA level, revealing a novel HCV-mediated control of IFN induction at the level of translation. The cellular protein kinase PKR is an important regulator of translation, through the phosphorylation of its substrate the eIF2alpha initiation factor. A comparison of the expression of luciferase placed under the control of an eIF2alpha-dependent (IRES(EMCV)) or independent (IRES(HCV)) RNA showed a specific HCV-mediated inhibition of eIF2alpha-dependent translation. We demonstrated that HCV infection triggers the phosphorylation of both PKR and eIF2alpha at 12 and 15 hrs post-infection. PKR silencing, as well as treatment with PKR pharmacological inhibitors, restored IFN induction in JFH1-infected cells, at least until 18 hrs post-infection, at which time a decrease in IFN expression could be attributed to NS3/4A-mediated MAVS cleavage. Importantly, both PKR silencing and PKR inhibitors led to inhibition of HCV yields in cells that express functional RIG-I/MAVS. In conclusion, here we provide the first evidence that HCV uses PKR to restrain its ability to induce IFN through the RIG-I/MAVS pathway. This opens up new possibilities to assay PKR chemical inhibitors for their potential to boost innate immunity in HCV infection.
Conflict of interest statement
Figures
References
-
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. - PubMed
-
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. - PubMed
-
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. - PubMed
-
- Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
