

Epigenetic changes (aberrant DNA methylation) in colorectal neoplasia
- PMID: 20485652
- PMCID: PMC2871659
- DOI: 10.5009/gnl.2007.1.1.1
Epigenetic changes (aberrant DNA methylation) in colorectal neoplasia
Abstract
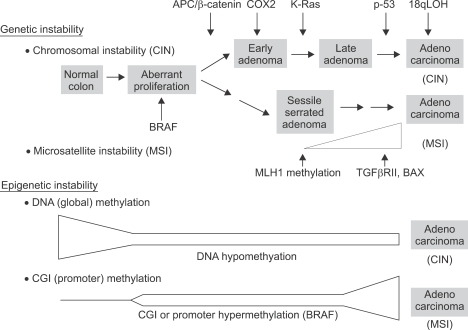
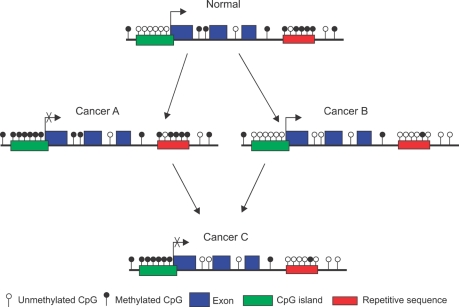
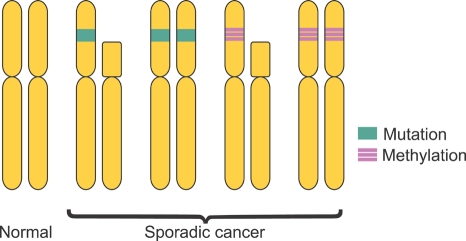
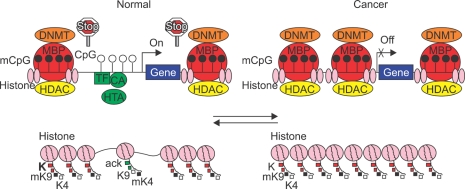
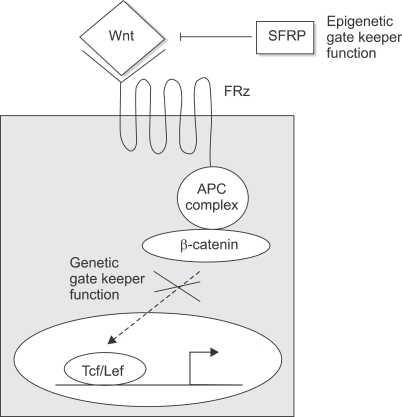
Both genetic and epigenetic events have been implicated in the stepwise histological progression involving adenoma-carcinoma and hyperplastic polyp/serrated adenoma-carcinoma sequences in the development of colorectal cancer. Genetic changes have been observed at each step in the initiation and progression of polyps to adenocarcinomas. Epigenetic changes also occur at each step in the pathogenesis of colorectal cancers and include CpG island DNA hypermethylation in the promoter region of genes resulting in transcriptional silencing through associated changes in chromatin structure and effects on binding of transcription factors, and DNA global hypomethylation which leads to chromosomal instability. Recent studies on MLH1 and APC genes indicate that epigenetic and genetic changes cooperate to facilitate tumor initiation and progression. Since aberrant CGI DNA promoter hypermethylation can be detected not only in colorectal polyps and cancers, but also in sera and stool, hypermethylated genes may serve as molecular markers for early detection, risk assessment and diagnosis. In addition, silenced genes caused by CGI DNA promoter hypermethylation can be reactivated by demethylating agents and also by both the inhibitors of DNA methyltransferases and histone deacetylases. Therefore, these epigenetically acting drugs should be evaluated for their chemopreventive and therapeutic potential for colorectal cancers.
Keywords: Colorectal cancer; Colorectal polyps; CpG island; DNA hypomethylation; DNA methylation; Epigenetic changes.
Figures
References
-
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. - PubMed
-
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. - PubMed
-
- Chung DC. The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis. Gastroenterology. 2000;119:854–865. - PubMed
-
- Rashid A, Issa JP. CpG island methylation in gastroenterologic neoplasia: a maturing field. Gastroenterology. 2004;127:1578–1588. - PubMed
-
- Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer and Metastasis. 2004;23:29–39. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
