

Current challenges of modeling diiron enzyme active sites for dioxygen activation by biomimetic synthetic complexes
- PMID: 20485834
- PMCID: PMC2909375
- DOI: 10.1039/c003079c
Current challenges of modeling diiron enzyme active sites for dioxygen activation by biomimetic synthetic complexes
Abstract
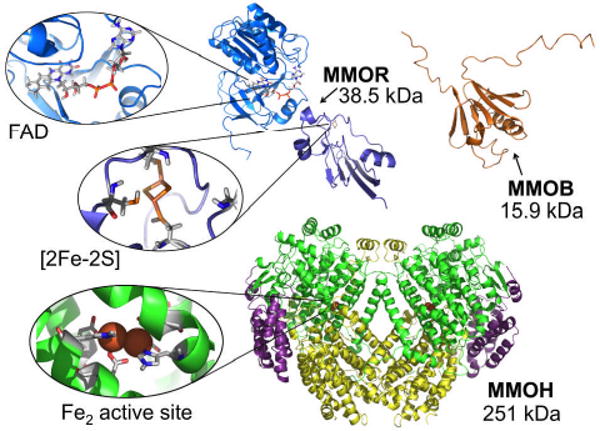
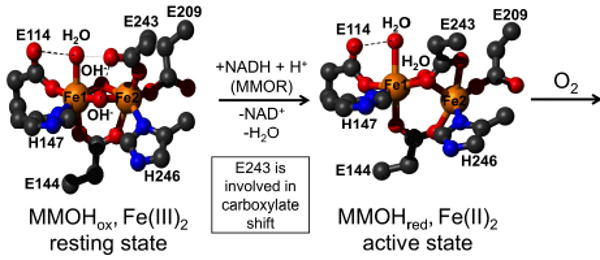
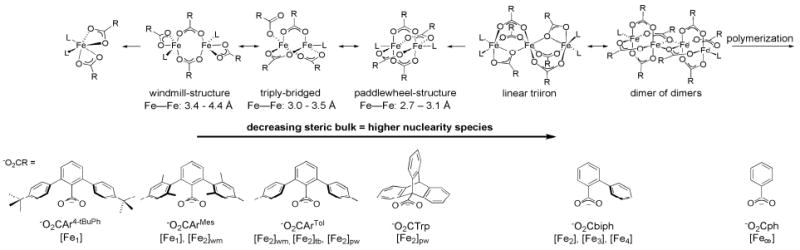
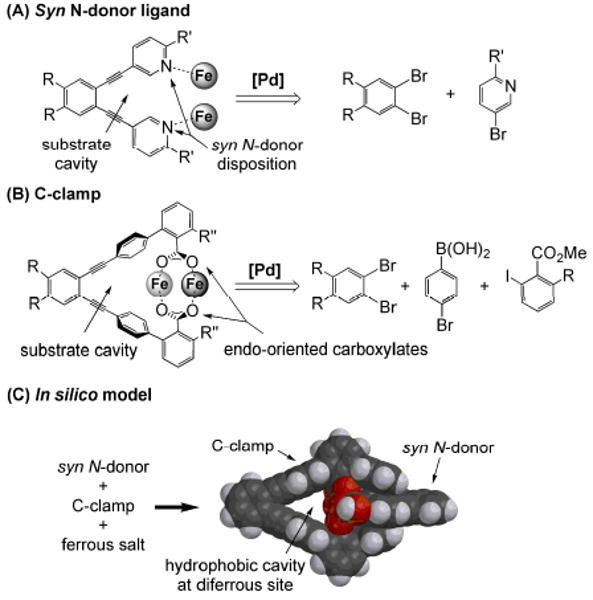
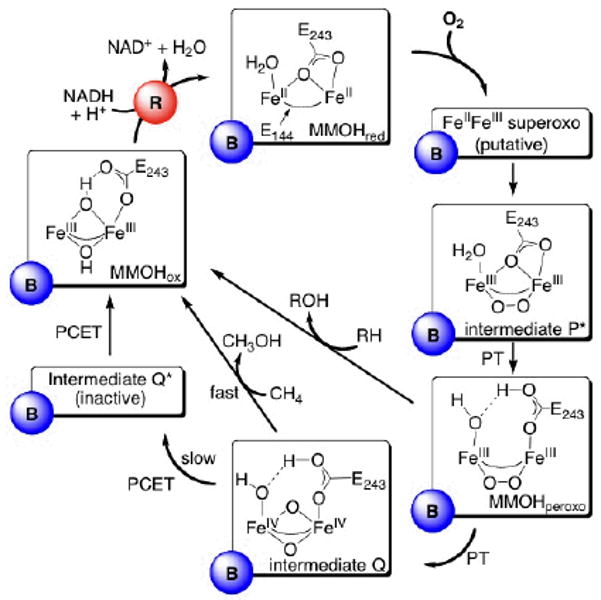
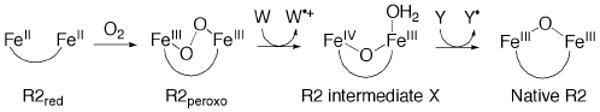
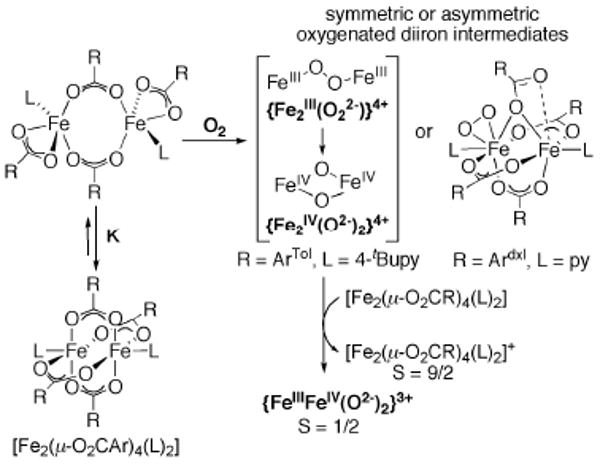
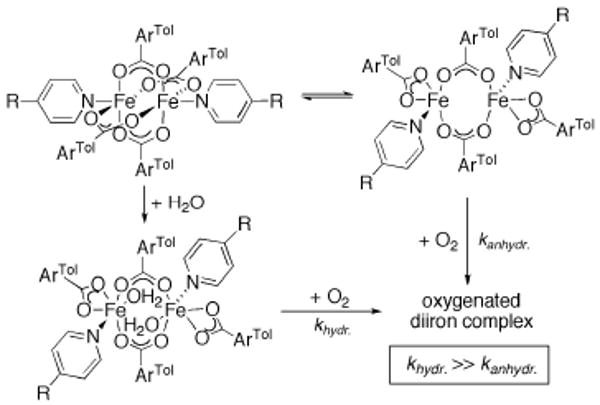
This tutorial review describes recent progress in modeling the active sites of carboxylate-rich non-heme diiron enzymes that activate dioxygen to carry out several key reactions in Nature. The chemistry of soluble methane monooxygenase, which catalyzes the selective oxidation of methane to methanol, is of particular interest for (bio)technological applications. Novel synthetic diiron complexes that mimic structural, and, to a lesser extent, functional features of these diiron enzymes are discussed. The chemistry of the enzymes is also briefly summarized. A particular focus of this review is on models that mimic characteristics of the diiron systems that were previously not emphasized, including systems that contain (i) aqua ligands, (ii) different substrates tethered to the ligand framework, (iii) dendrimers attached to carboxylates to mimic the protein environment, (iv) two N-donors in a syn-orientation with respect to the iron-iron vector, and (v) a N-rich ligand environment capable of accessing oxygenated high-valent diiron intermediates.
Figures
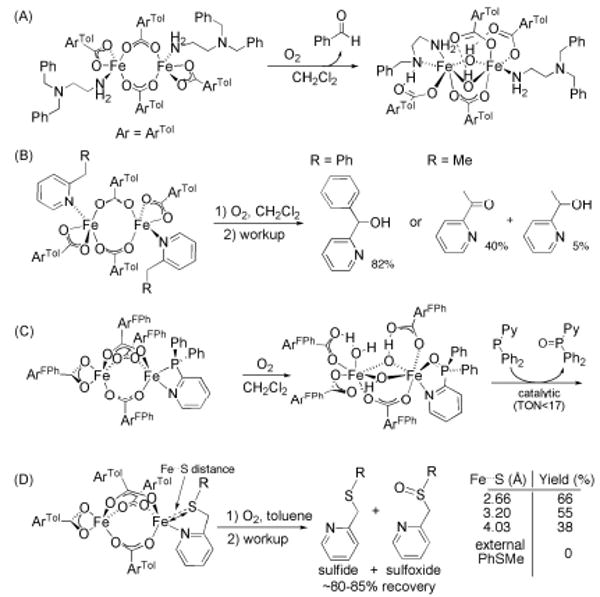
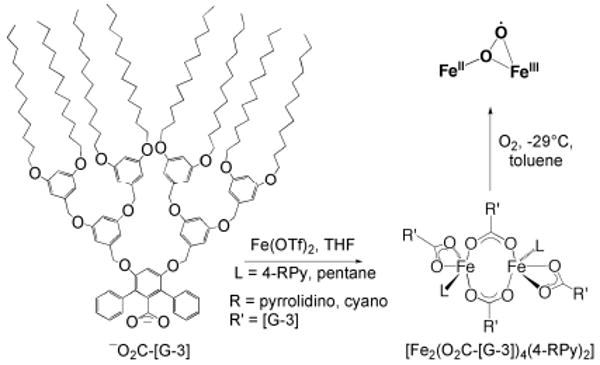
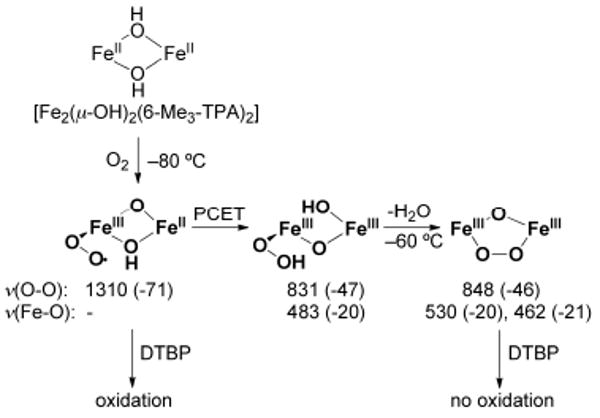
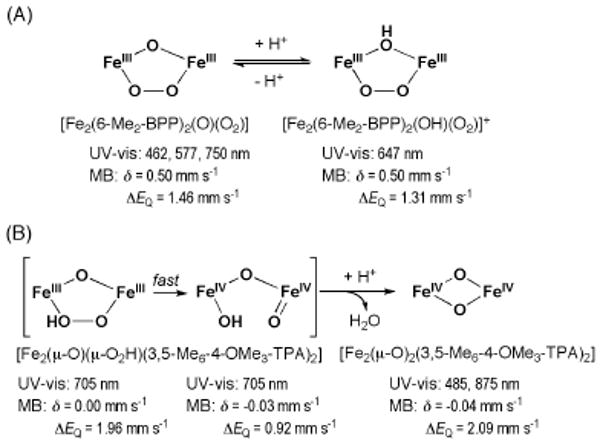
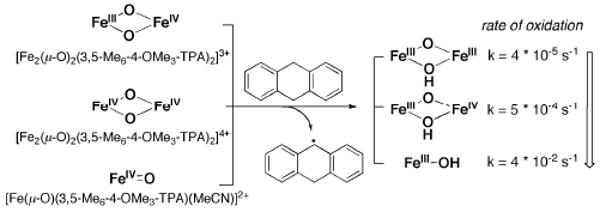
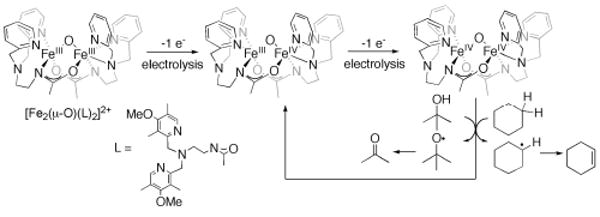
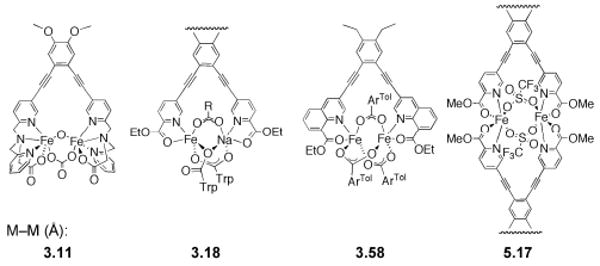
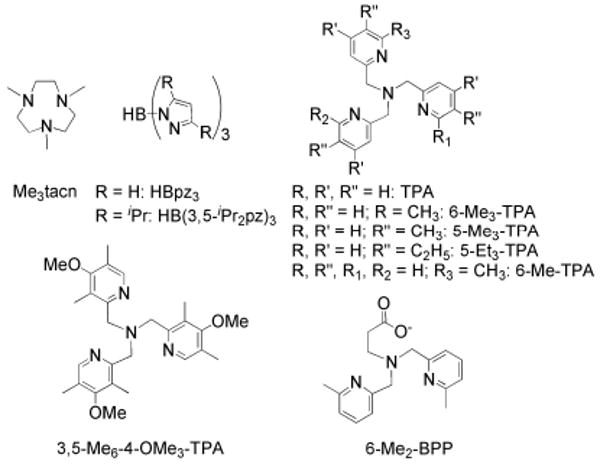
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous