

Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns
- PMID: 20488932
- PMCID: PMC2892098
- DOI: 10.1101/gr.101535.109
Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns
Abstract
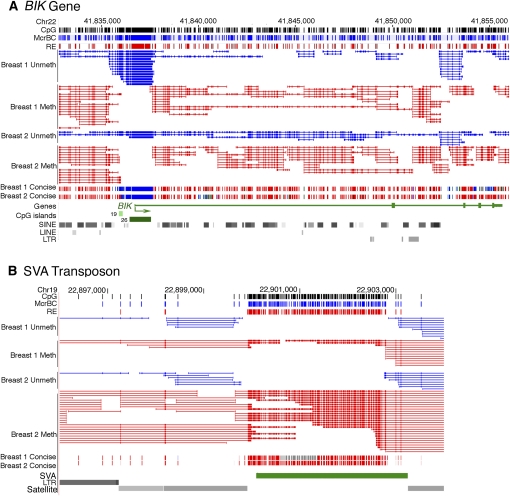
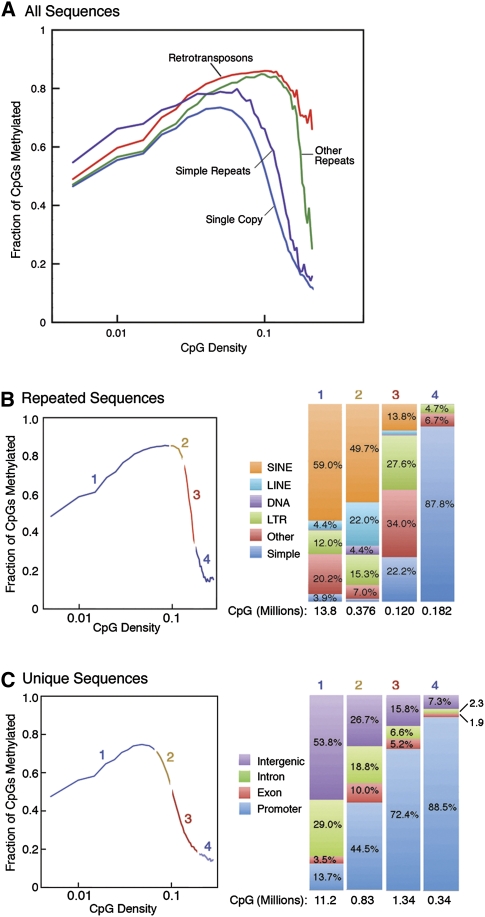
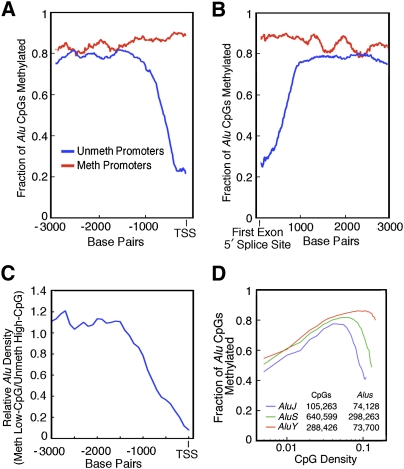
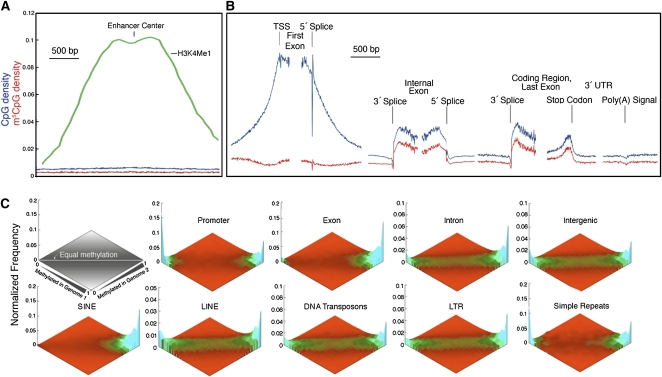
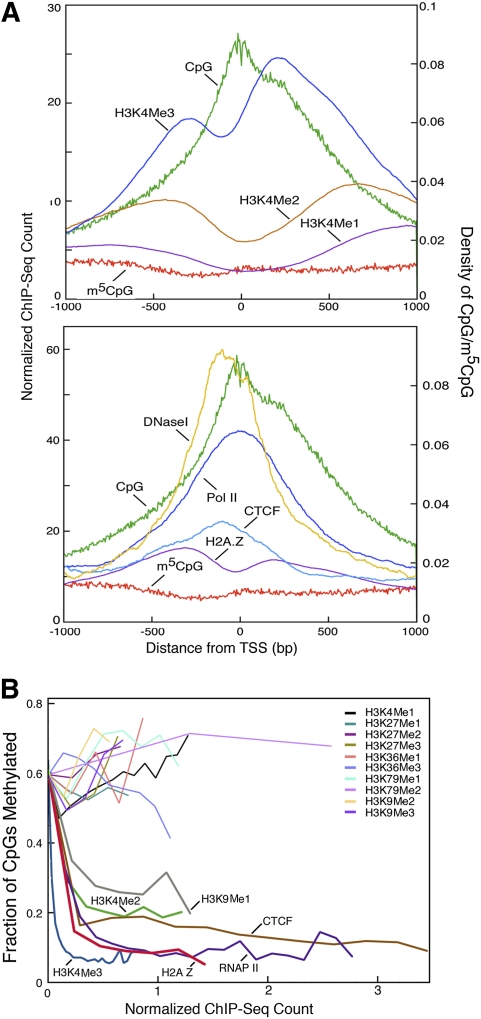
Abnormalities of genomic methylation patterns are lethal or cause disease, but the cues that normally designate CpG dinucleotides for methylation are poorly understood. We have developed a new method of methylation profiling that has single-CpG resolution and can address the methylation status of repeated sequences. We have used this method to determine the methylation status of >275 million CpG sites in human and mouse DNA from breast and brain tissues. Methylation density at most sequences was found to increase linearly with CpG density and to fall sharply at very high CpG densities, but transposons remained densely methylated even at higher CpG densities. The presence of histone H2A.Z and histone H3 di- or trimethylated at lysine 4 correlated strongly with unmethylated DNA and occurred primarily at promoter regions. We conclude that methylation is the default state of most CpG dinucleotides in the mammalian genome and that a combination of local dinucleotide frequencies, the interaction of repeated sequences, and the presence or absence of histone variants or modifications shields a population of CpG sites (most of which are in and around promoters) from DNA methyltransferases that lack intrinsic sequence specificity.
Figures
References
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–830 - PubMed
-
- Bell AC, Felsenfeld G 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405: 482–488 - PubMed
-
- Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases