

Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene
- PMID: 20493458
- PMCID: PMC3032067
- DOI: 10.1016/j.ajhg.2010.04.012
Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene
Abstract
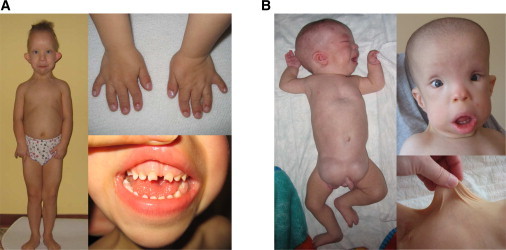
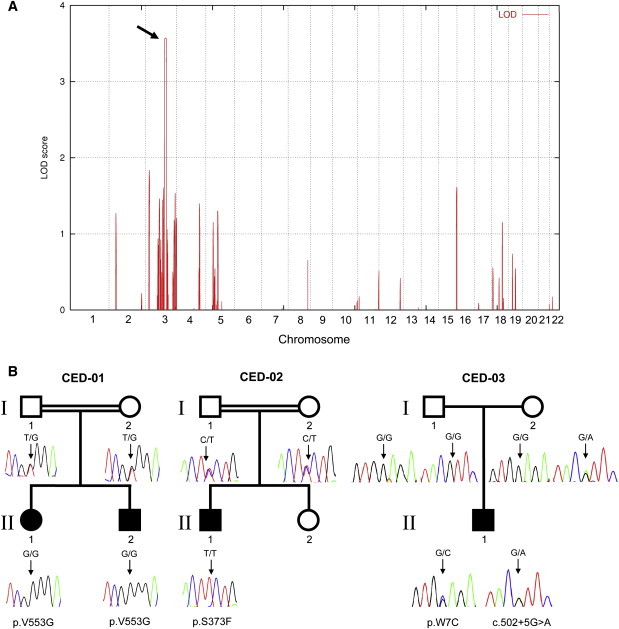
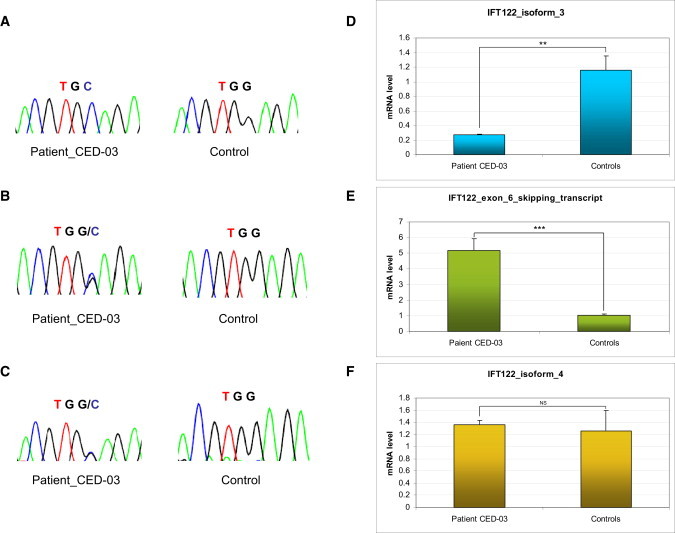
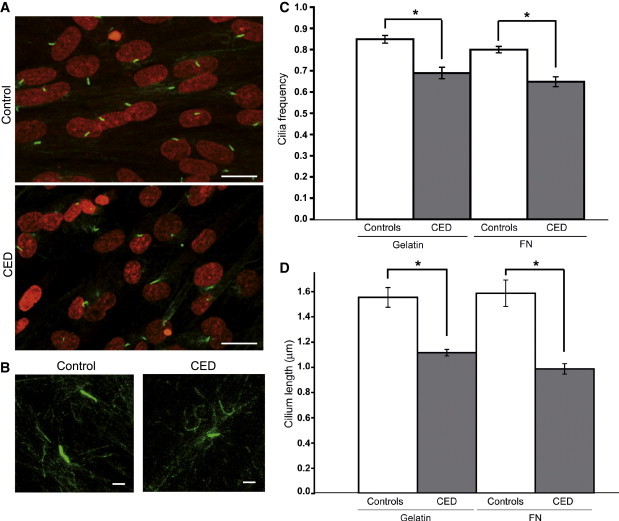
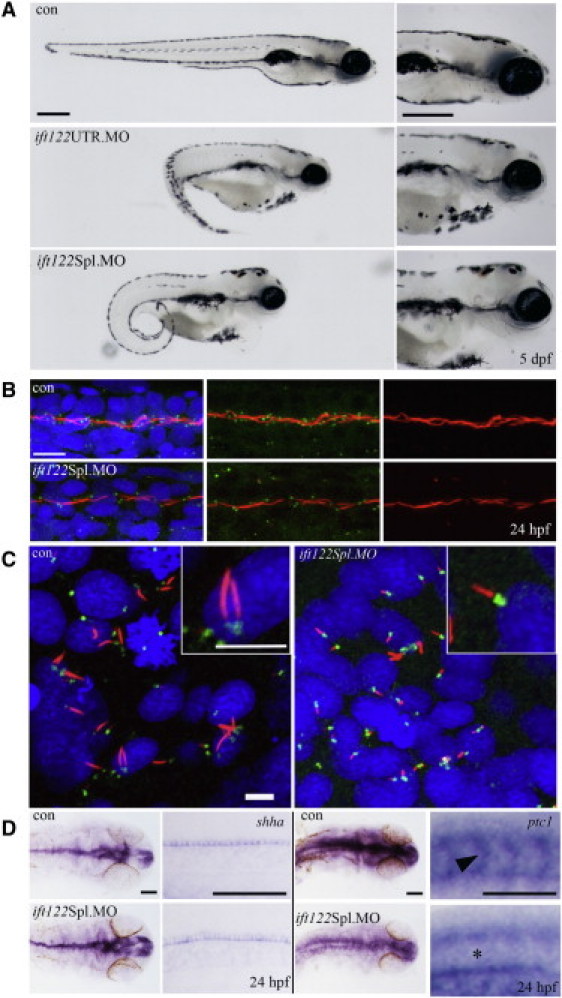
Cranioectodermal dysplasia (CED) is a disorder characterized by craniofacial, skeletal, and ectodermal abnormalities. Most cases reported to date are sporadic, but a few familial cases support an autosomal-recessive inheritance pattern. Aiming at the elucidation of the genetic basis of CED, we collected 13 patients with CED symptoms from 12 independent families. In one family with consanguineous parents two siblings were affected, permitting linkage analysis and homozygosity mapping. This revealed a single region of homozygosity with a significant LOD score (3.57) on chromosome 3q21-3q24. By sequencing candidate genes from this interval we found a homozygous missense mutation in the IFT122 (WDR10) gene that cosegregated with the disease. Examination of IFT122 in our patient cohort revealed one additional homozygous missense change in the patient from a second consanguineous family. In addition, we found compound heterozygosity for a donor splice-site change and a missense change in one sporadic patient. All mutations were absent in 340 control chromosomes. Because IFT122 plays an important role in the assembly and maintenance of eukaryotic cilia, we investigated patient fibroblasts and found significantly reduced frequency and length of primary cilia as compared to controls. Furthermore, we transiently knocked down ift122 in zebrafish embryos and observed the typical phenotype found in other models of ciliopathies. Because not all of our patients harbored mutations in IFT122, CED seems to be genetically heterogeneous. Still, by identifying CED as a ciliary disorder, our study suggests that the causative mutations in the unresolved cases most likely affect primary cilia function too.
Copyright 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. - PubMed
-
- Konstantinidou A.E., Fryssira H., Sifakis S., Karadimas C., Kaminopetros P., Agrogiannis G., Velonis S., Nikkels P.G., Patsouris E. Cranioectodermal dysplasia: A probable ciliopathy. Am. J. Med. Genet. A. 2009;149A:2206–2211. - PubMed
-
- Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
