

Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability
- PMID: 20493460
- PMCID: PMC3032071
- DOI: 10.1016/j.ajhg.2010.05.001
Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability
Abstract
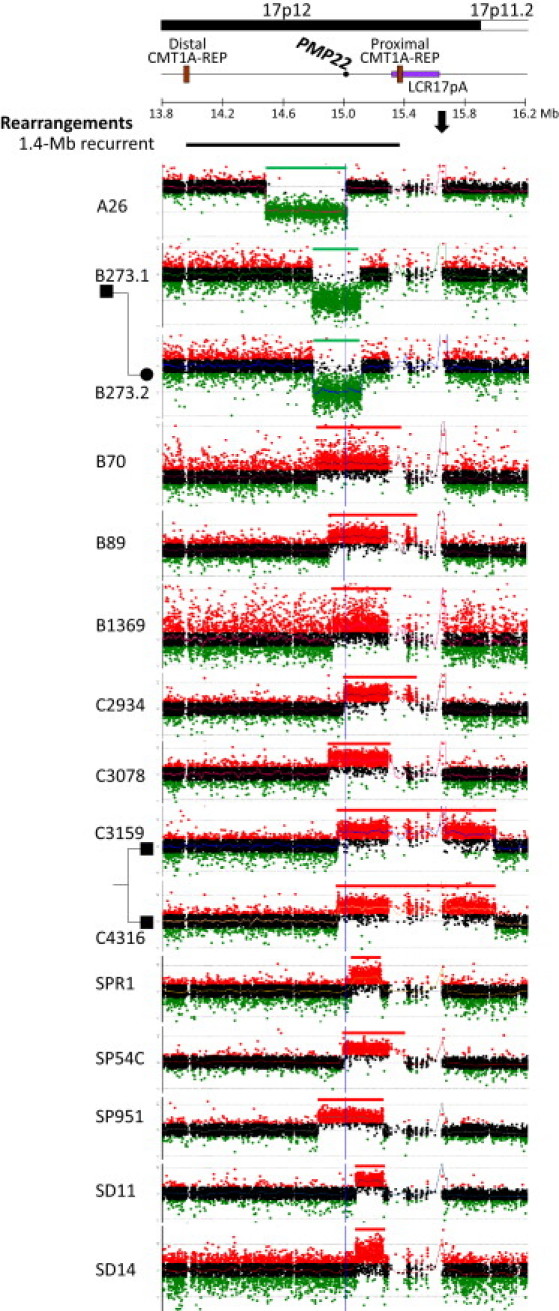
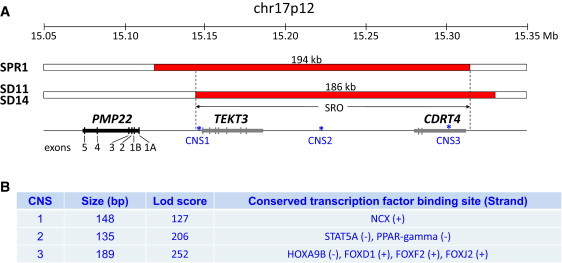
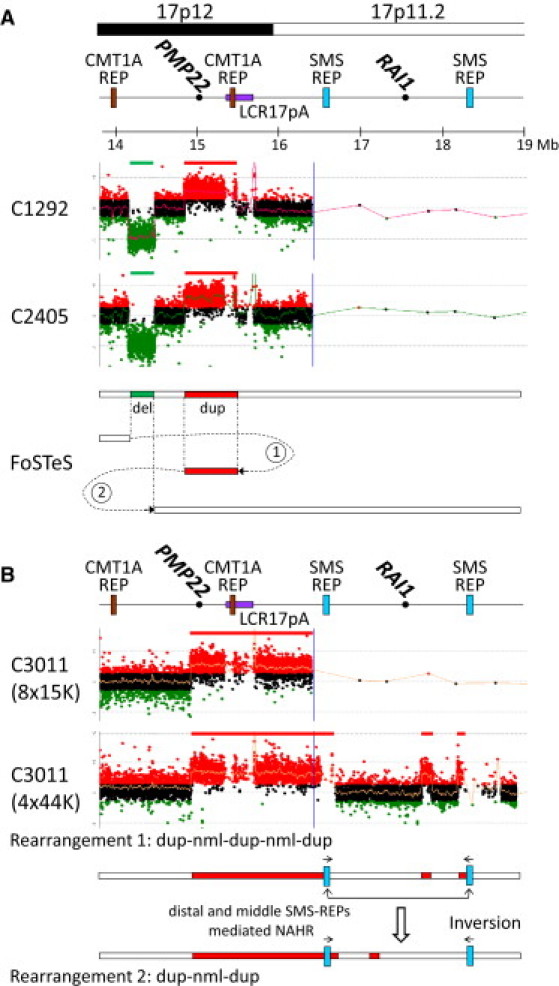
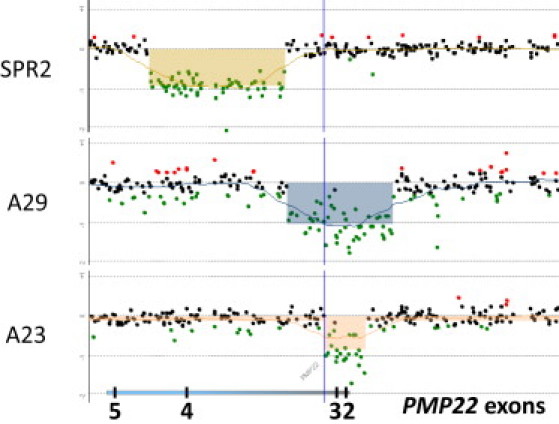
Genomic rearrangements involving the peripheral myelin protein gene (PMP22) in human chromosome 17p12 are associated with neuropathy: duplications cause Charcot-Marie-Tooth disease type 1A (CMT1A), whereas deletions lead to hereditary neuropathy with liability to pressure palsies (HNPP). Our previous studies showed that >99% of these rearrangements are recurrent and mediated by nonallelic homologous recombination (NAHR). Rare copy number variations (CNVs) generated by nonrecurrent rearrangements also exist in 17p12, but their underlying mechanisms are not well understood. We investigated 21 subjects with rare CNVs associated with CMT1A or HNPP by oligonucleotide-based comparative genomic hybridization microarrays and breakpoint sequence analyses, and we identified 17 unique CNVs, including two genomic deletions, ten genomic duplications, two complex rearrangements, and three small exonic deletions. Each of these CNVs includes either the entire PMP22 gene, or exon(s) only, or ultraconserved potential regulatory sequences upstream of PMP22, further supporting the contention that PMP22 is the critical gene mediating the neuropathy phenotypes associated with 17p12 rearrangements. Breakpoint sequence analysis reveals that, different from the predominant NAHR mechanism in recurrent rearrangement, various molecular mechanisms, including nonhomologous end joining, Alu-Alu-mediated recombination, and replication-based mechanisms (e.g., FoSTeS and/or MMBIR), can generate nonrecurrent 17p12 rearrangements associated with neuropathy. We document a multitude of ways in which gene function can be altered by CNVs. Given the characteristics, including small size, structural complexity, and location outside of coding regions, of selected rare CNVs, their identification remains a challenge for genome analysis. Rare CNVs may potentially represent an important portion of "missing heritability" for human diseases.
Copyright 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Nonrecurrent 17p11.2p12 Rearrangement Events that Result in Two Concomitant Genomic Disorders: The PMP22-RAI1 Contiguous Gene Duplication Syndrome.Am J Hum Genet. 2015 Nov 5;97(5):691-707. doi: 10.1016/j.ajhg.2015.10.003. Am J Hum Genet. 2015. PMID: 26544804 Free PMC article.
-
Inheritance of Charcot-Marie-Tooth disease 1A with rare nonrecurrent genomic rearrangement.Neurogenetics. 2011 Feb;12(1):51-8. doi: 10.1007/s10048-010-0272-3. Epub 2010 Dec 31. Neurogenetics. 2011. PMID: 21193943
-
The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes.Genome Res. 2001 Jun;11(6):1018-33. doi: 10.1101/gr.180401. Genome Res. 2001. PMID: 11381029 Free PMC article.
-
Charcot-Marie-Tooth disease and related inherited neuropathies.Medicine (Baltimore). 1996 Sep;75(5):233-50. doi: 10.1097/00005792-199609000-00001. Medicine (Baltimore). 1996. PMID: 8862346 Review.
-
PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and Hereditary Neuropathy with liability to Pressure Palsies.Orphanet J Rare Dis. 2014 Mar 19;9:38. doi: 10.1186/1750-1172-9-38. Orphanet J Rare Dis. 2014. PMID: 24646194 Free PMC article. Review.
Cited by
-
A 3.4-kb Copy-Number Deletion near EPAS1 Is Significantly Enriched in High-Altitude Tibetans but Absent from the Denisovan Sequence.Am J Hum Genet. 2015 Jul 2;97(1):54-66. doi: 10.1016/j.ajhg.2015.05.005. Epub 2015 Jun 11. Am J Hum Genet. 2015. PMID: 26073780 Free PMC article.
-
Variation in SIPA1L2 is correlated with phenotype modification in Charcot- Marie- Tooth disease type 1A.Ann Neurol. 2019 Mar;85(3):316-330. doi: 10.1002/ana.25426. Ann Neurol. 2019. PMID: 30706531 Free PMC article.
-
Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer.Hum Mol Genet. 2018 Aug 15;27(16):2830-2839. doi: 10.1093/hmg/ddy191. Hum Mol Genet. 2018. PMID: 29771329 Free PMC article.
-
Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability.BMC Med Genet. 2015 Sep 21;16:84. doi: 10.1186/s12881-015-0224-8. BMC Med Genet. 2015. PMID: 26392352 Free PMC article.
-
Comparison between clinical disabilities and electrophysiological values in Charcot-Marie-Tooth 1A patients with PMP22 duplication.J Clin Neurol. 2012 Jun;8(2):139-45. doi: 10.3988/jcn.2012.8.2.139. Epub 2012 Jun 29. J Clin Neurol. 2012. PMID: 22787498 Free PMC article.
References
-
- Lupski J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. - PubMed
-
- Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. - PubMed
-
- Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. - PubMed
-
- Chance P.F., Alderson M.K., Leppig K.A., Lensch M.W., Matsunami N., Smith B., Swanson P.D., Odelberg S.J., Disteche C.M., Bird T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
