

Dyskeratosis congenita
- PMID: 20493861
- PMCID: PMC3238451
- DOI: 10.1016/j.febslet.2010.05.019
Dyskeratosis congenita
Abstract
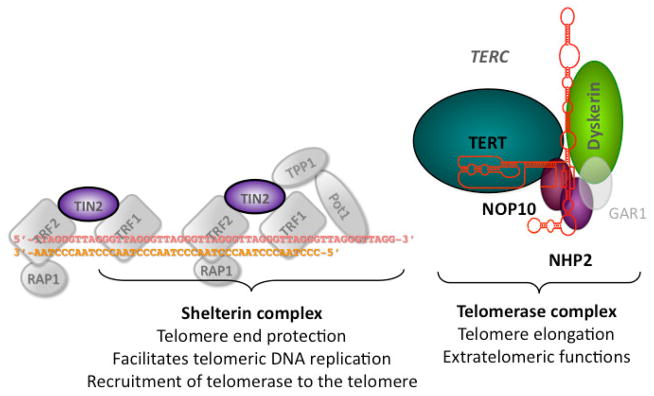
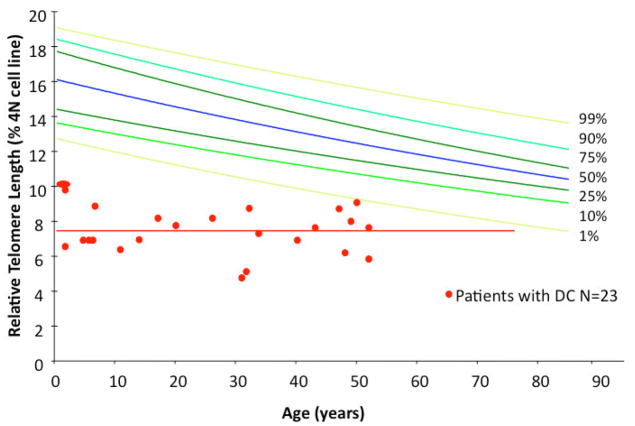
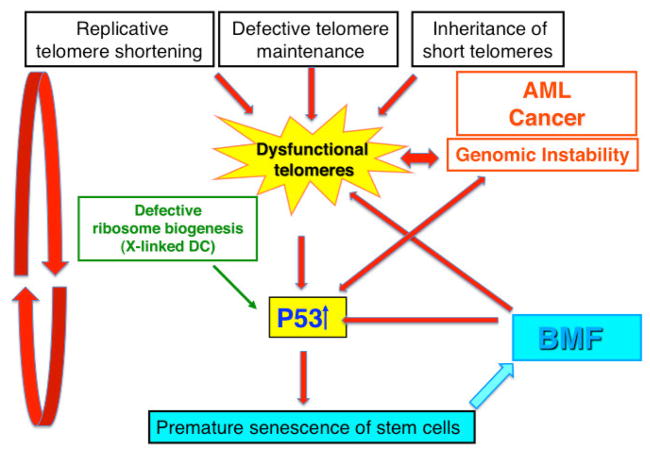
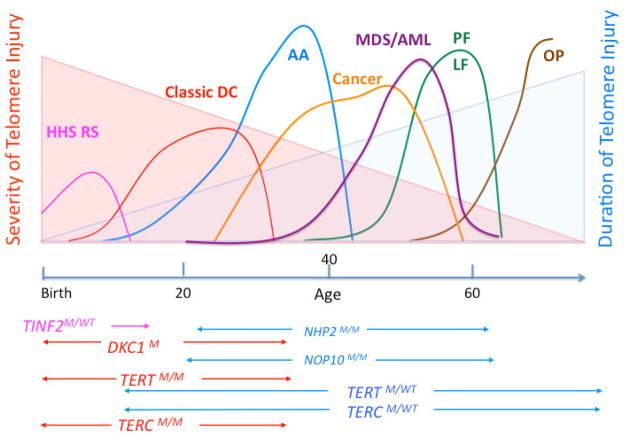
Dyskeratosis congenita (DC) was originally defined as a rare inherited bone marrow failure (BMF) syndrome associated with distinct mucocutaneous features. Today DC is defined by its pathogenetic mechanism and mutations in components of the telomere maintenance machinery resulting in excessively short telomeres in highly proliferating tissues. With this new definition the disease spectrum has broadened and ranges from intrauterine growth retardation, cerebellar hypoplasia, and death in early childhood to asymptomatic mutation carriers whose descendants are predisposed to malignancy, BMF, or pulmonary disease. The degree of telomere dysfunction is the major determinant of disease onset and manifestations.
Copyright 2010 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Bessler M, Mason P, Link D, Wilson D. Nathan and Oski’s Hematology of Infancy and Childhood. In: Orkin S, Nathan D, Ginsburg D, Look A, Fisher D, Lux S, editors. Saunders; Philadelphia: 2008. pp. 307–395.
-
- Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
