

cAMP mediators of pulsatile insulin secretion from glucose-stimulated single beta-cells
- PMID: 20498366
- PMCID: PMC2906294
- DOI: 10.1074/jbc.M109.095992
cAMP mediators of pulsatile insulin secretion from glucose-stimulated single beta-cells
Abstract
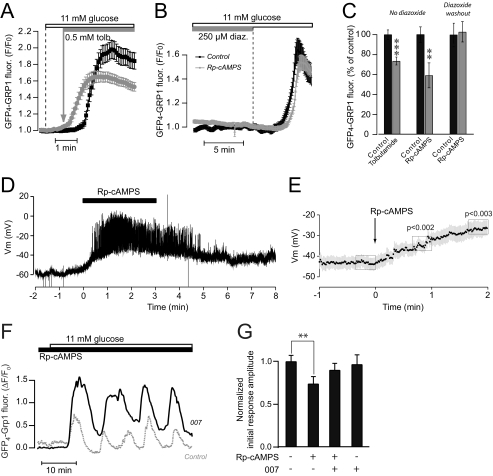
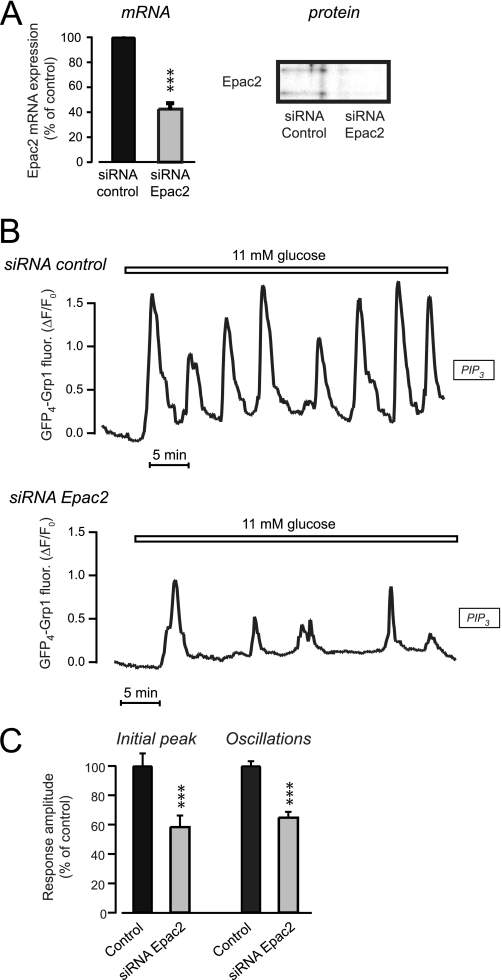
Pulsatile insulin release from glucose-stimulated beta-cells is driven by oscillations of the Ca(2+) and cAMP concentrations in the subplasma membrane space ([Ca(2+)](pm) and [cAMP](pm)). To clarify mechanisms by which cAMP regulates insulin secretion, we performed parallel evanescent wave fluorescence imaging of [cAMP](pm), [Ca(2+)](pm), and phosphatidylinositol 3,4,5-trisphosphate (PIP(3)) in the plasma membrane. This lipid is formed by autocrine insulin receptor activation and was used to monitor insulin release kinetics from single MIN6 beta-cells. Elevation of the glucose concentration from 3 to 11 mm induced, after a 2.7-min delay, coordinated oscillations of [Ca(2+)](pm), [cAMP](pm), and PIP(3). Inhibitors of protein kinase A (PKA) markedly diminished the PIP(3) response when applied before glucose stimulation, but did not affect already manifested PIP(3) oscillations. The reduced PIP(3) response could be attributed to accelerated depolarization causing early rise of [Ca(2+)](pm) that preceded the elevation of [cAMP](pm). However, the amplitude of the PIP(3) response after PKA inhibition was restored by a specific agonist to the cAMP-dependent guanine nucleotide exchange factor Epac. Suppression of cAMP formation with adenylyl cyclase inhibitors reduced already established PIP(3) oscillations in glucose-stimulated cells, and this effect was almost completely counteracted by the Epac agonist. In cells treated with small interfering RNA targeting Epac2, the amplitudes of the glucose-induced PIP(3) oscillations were reduced, and the Epac agonist was without effect. The data indicate that temporal coordination of the triggering [Ca(2+)](pm) and amplifying [cAMP](pm) signals is important for glucose-induced pulsatile insulin release. Although both PKA and Epac2 partake in initiating insulin secretion, the cAMP dependence of established pulsatility is mediated by Epac2.
Figures





References
-
- Henquin J. C. (2000) Diabetes 49, 1751–1760 - PubMed
-
- Tengholm A., Gylfe E. (2009) Mol. Cell. Endocrinol. 297, 58–72 - PubMed
-
- Henquin J. C. (2009) Diabetologia 52, 739–751 - PubMed
-
- Bratanova-Tochkova T. K., Cheng H., Daniel S., Gunawardana S., Liu Y. J., Mulvaney-Musa J., Schermerhorn T., Straub S. G., Yajima H., Sharp G. W. (2002) Diabetes 51, Suppl. 1, S83–S90 - PubMed
-
- Rorsman P., Eliasson L., Renström E., Gromada J., Barg S., Göpel S. (2000) News Physiol. Sci. 15, 72–77 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
