

Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of peroxisome proliferator-activated receptor gamma activation in mice
- PMID: 20498376
- PMCID: PMC2911274
- DOI: 10.1074/jbc.M110.123174
Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of peroxisome proliferator-activated receptor gamma activation in mice
Abstract
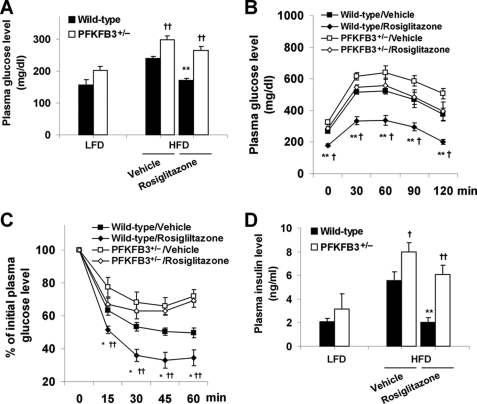
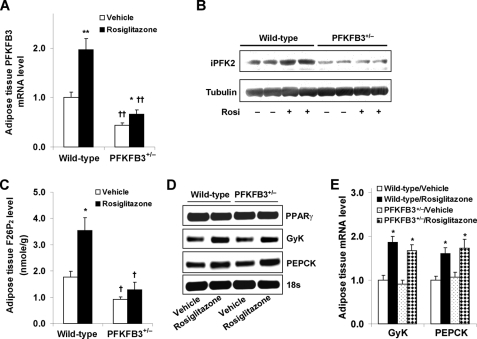
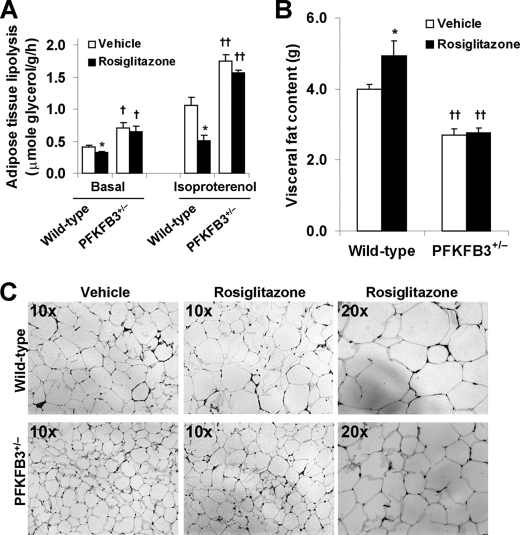
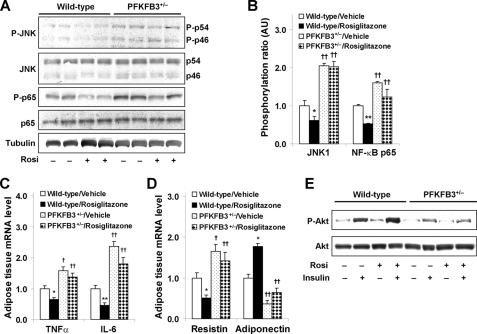
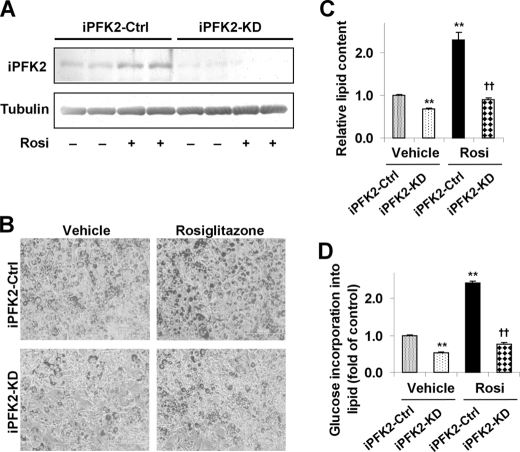
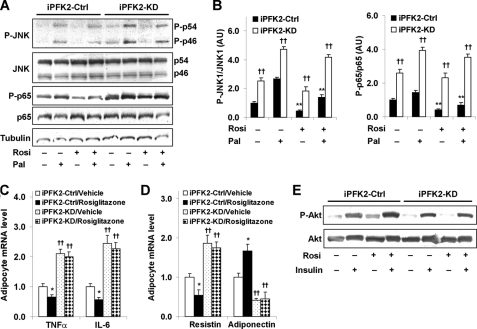
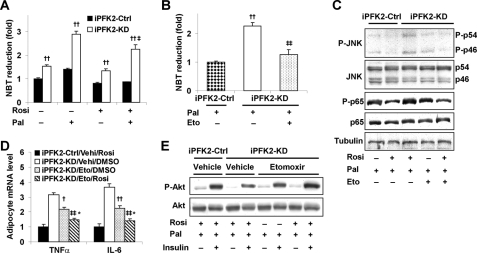
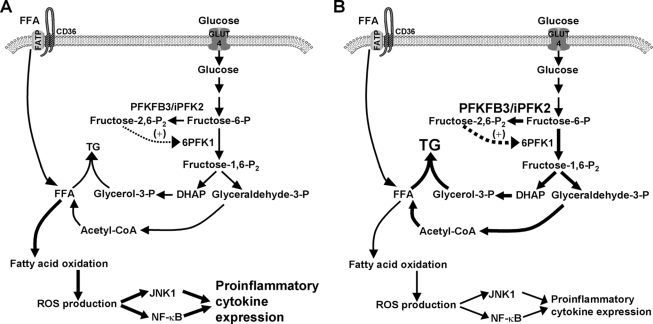
PFKFB3 is the gene that codes for the inducible isoform of 6-phosphofructo-2-kinase (iPFK2), a key regulatory enzyme of glycolysis. As one of the targets of peroxisome proliferator-activated receptor gamma (PPARgamma), PFKFB3/iPFK2 is up-regulated by thiazolidinediones. In the present study, using PFKFB3/iPFK2-disrupted mice, the role of PFKFB3/iPFK2 in the anti-diabetic effect of PPARgamma activation was determined. In wild-type littermate mice, PPARgamma activation (i.e. treatment with rosiglitazone) restored euglycemia and reversed high fat diet-induced insulin resistance and glucose intolerance. In contrast, PPARgamma activation did not reduce high fat diet-induced hyperglycemia and failed to reverse insulin resistance and glucose intolerance in PFKFB3(+/-) mice. The lack of anti-diabetic effect in PFKFB3(+/-) mice was associated with the inability of PPARgamma activation to suppress adipose tissue lipolysis and proinflammatory cytokine production, stimulate visceral fat accumulation, enhance adipose tissue insulin signaling, and appropriately regulate adipokine expression. Similarly, in cultured 3T3-L1 adipocytes, knockdown of PFKFB3/iPFK2 lessened the effect of PPARgamma activation on stimulating lipid accumulation. Furthermore, PPARgamma activation did not suppress inflammatory signaling in PFKFB3/iPFK2-knockdown adipocytes as it did in control adipocytes. Upon inhibition of excessive fatty acid oxidation in PFKFB3/iPFK2-knockdown adipocytes, PPARgamma activation was able to significantly reverse inflammatory signaling and proinflammatory cytokine expression and restore insulin signaling. Together, these data demonstrate that PFKFB3/iPFK2 is critically involved in the anti-diabetic effect of PPARgamma activation.
Figures
Similar articles
-
Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses.J Biol Chem. 2012 Jun 15;287(25):21492-500. doi: 10.1074/jbc.M112.370379. Epub 2012 May 3. J Biol Chem. 2012. PMID: 22556414 Free PMC article.
-
Disruption of inducible 6-phosphofructo-2-kinase impairs the suppressive effect of PPARγ activation on diet-induced intestine inflammatory response.J Nutr Biochem. 2013 May;24(5):770-5. doi: 10.1016/j.jnutbio.2012.04.007. Epub 2012 Jul 26. J Nutr Biochem. 2013. PMID: 22841546 Free PMC article.
-
Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response.J Biol Chem. 2010 Feb 5;285(6):3713-3721. doi: 10.1074/jbc.M109.058446. Epub 2009 Nov 30. J Biol Chem. 2010. PMID: 19948719 Free PMC article.
-
Review: Peroxisome proliferator-activated receptor gamma and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose metabolism.J Clin Endocrinol Metab. 2007 Feb;92(2):386-95. doi: 10.1210/jc.2006-1268. Epub 2006 Dec 5. J Clin Endocrinol Metab. 2007. PMID: 17148564 Review.
-
Controlling a master switch of adipocyte development and insulin sensitivity: covalent modifications of PPARγ.Biochim Biophys Acta. 2012 Jul;1822(7):1090-5. doi: 10.1016/j.bbadis.2012.03.014. Epub 2012 Apr 4. Biochim Biophys Acta. 2012. PMID: 22504298 Free PMC article. Review.
Cited by
-
Glucose and Palmitate Differentially Regulate PFKFB3/iPFK2 and Inflammatory Responses in Mouse Intestinal Epithelial Cells.Sci Rep. 2016 Jul 8;6:28963. doi: 10.1038/srep28963. Sci Rep. 2016. PMID: 27387960 Free PMC article.
-
Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses.J Biol Chem. 2012 Jun 15;287(25):21492-500. doi: 10.1074/jbc.M112.370379. Epub 2012 May 3. J Biol Chem. 2012. PMID: 22556414 Free PMC article.
-
PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT.Cell Death Dis. 2018 Apr 1;9(4):428. doi: 10.1038/s41419-018-0435-y. Cell Death Dis. 2018. PMID: 29559632 Free PMC article.
-
The many ways tissue phagocytes respond to dying cells.Immunol Rev. 2017 May;277(1):158-173. doi: 10.1111/imr.12537. Immunol Rev. 2017. PMID: 28462530 Free PMC article. Review.
-
Disruption of inducible 6-phosphofructo-2-kinase impairs the suppressive effect of PPARγ activation on diet-induced intestine inflammatory response.J Nutr Biochem. 2013 May;24(5):770-5. doi: 10.1016/j.jnutbio.2012.04.007. Epub 2012 Jul 26. J Nutr Biochem. 2013. PMID: 22841546 Free PMC article.
References
-
- Lehmann J. M., Moore L. B., Smith-Oliver T. A., Wilkison W. O., Willson T. M., Kliewer S. A. (1995) J. Biol. Chem. 270, 12953–12956 - PubMed
-
- Berger J., Bailey P., Biswas C., Cullinan C. A., Doebber T. W., Hayes N. S., Saperstein R., Smith R. G., Leibowitz M. D. (1996) Endocrinology 137, 4189–4195 - PubMed
-
- Lebovitz H. E., Dole J. F., Patwardhan R., Rappaport E. B., Freed M. I. (2001) J. Clin. Endocrinol. Metab. 86, 280–288 - PubMed
-
- Phillips L. S., Grunberger G., Miller E., Patwardhan R., Rappaport E. B., Salzman A. (2001) Diabetes Care 24, 308–315 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
