

Dysosteosclerosis presents as an "osteoclast-poor" form of osteopetrosis: comprehensive investigation of a 3-year-old girl and literature review
- PMID: 20499338
- PMCID: PMC3179286
- DOI: 10.1002/jbmr.131
Dysosteosclerosis presents as an "osteoclast-poor" form of osteopetrosis: comprehensive investigation of a 3-year-old girl and literature review
Abstract
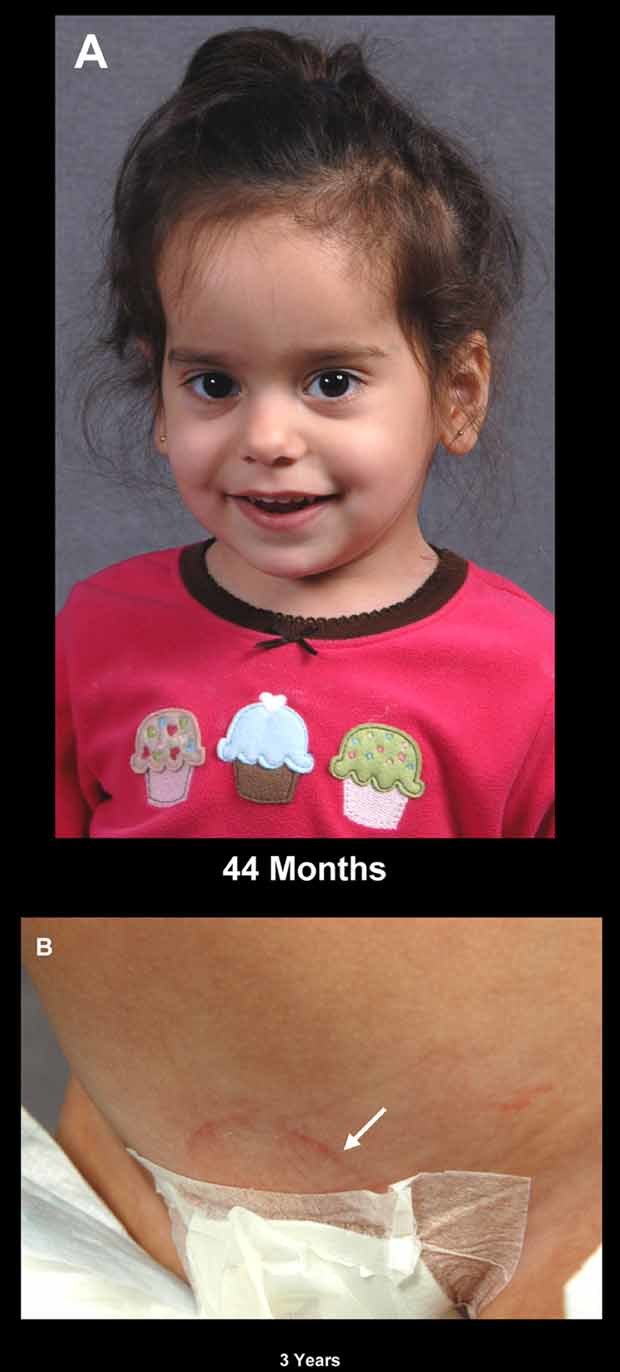
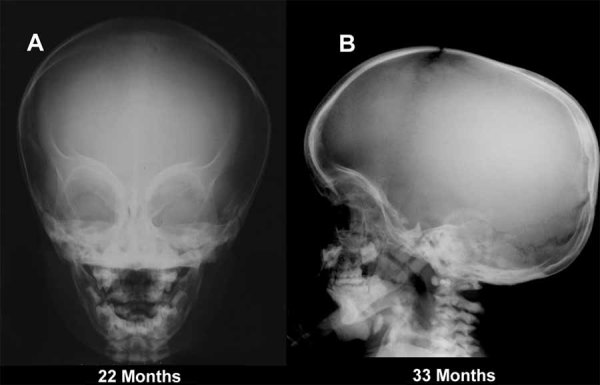
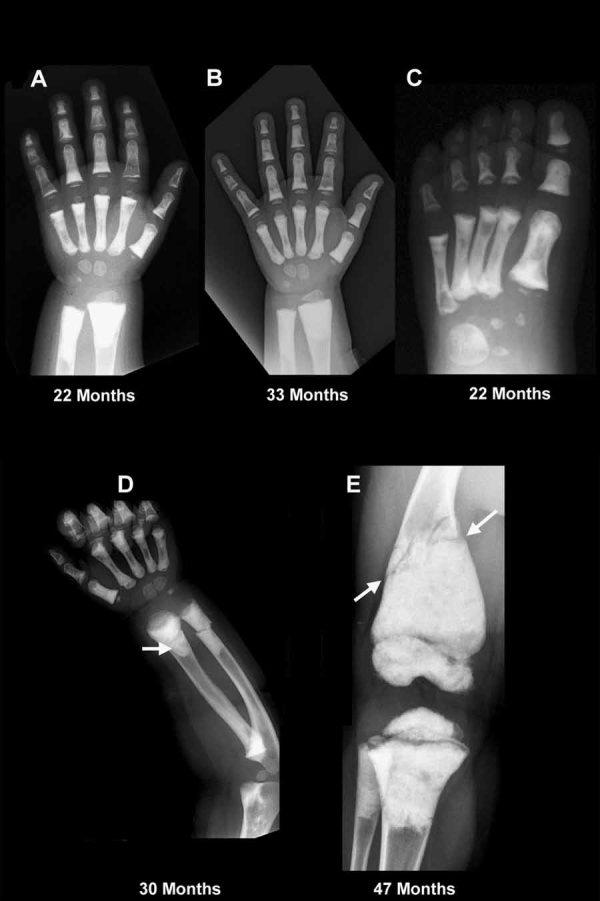
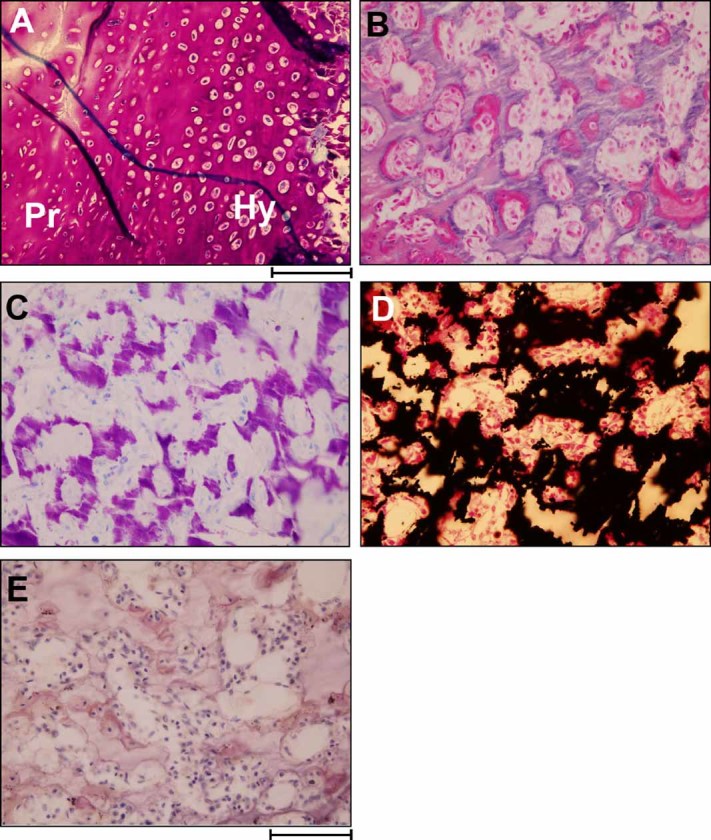
Dysosteosclerosis (DSS), an extremely rare dense bone disease, features short stature and fractures and sometimes optic atrophy, cranial nerve palsy, developmental delay, and failure of tooth eruption in infancy or early childhood consistent with osteopetrosis (OPT). Bone histology during childhood shows unresorbed primary spongiosa from deficient osteoclast action. Additionally, there is remarkable progressive flattening of all vertebrae and, by adolescence, paradoxical metaphyseal osteopenia with thin cortical bone. Reports of consanguinity indicate autosomal recessive inheritance, yet more affected males than females suggest X-linked recessive inheritance. We investigated a nonconsanguineous girl with DSS. Osteosclerosis was discovered at age 7 months. Our studies, spanning ages 11 to 44 months, showed weight at approximately 50th percentile, and length diminishing from approximately 30th percentile to -2.3 SD. Head circumference was +4 SD. The patient had frontal bossing, blue sclera, normal teeth, genu valgum, and unremarkable joints. Radiographs showed orbital and facial sclerosis, basilar thickening, bone-in-bone appearance of the pelvis, sclerotic long bone ends, and fractures of ribs and extremities. Progressive metaphyseal widening occurred as vertebrae changed from ovoid to flattened and became beaked anteriorly. A hemogram was normal. Consistent with OPT, serum parathyroid hormone (PTH) concentrations reflected dietary calcium levels. Serum bone alkaline phosphatase, osteocalcin, and TRACP-5b were subnormal. The iliac crest contained excessive primary spongiosa and no osteoclasts. No mutations were identified in the splice sites or exons for the genes encoding chloride channel 7, T-cell immune regulator 1, OPT-associated transmembrane protein 1, and monocyte colony-stimulating factor (M-CSF) and its receptor C-FMS, ANKH, OPG, RANK, and RANKL. Genomic copy-number microarray was unrevealing. Hence, DSS is a distinctive OPT of unknown etiology featuring osteoclast deficiency during early childhood. How osteopenia follows is an enigma of human skeletal pathobiology.
© 2010 American Society for Bone and Mineral Research.
Figures


References
-
- Online Mendelian Inheritance in Man (OMIM) McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), November 11, 2009; http://www.ncbi.nlm.nih.gov/omim/
-
- Spranger J, Albrecht C, Rohwedder HJ, Wiedemann HR. Dysosteosclerosis: a special form of generalized osteosclerosis. Fortschr Geb Rontgenstr Nuklearmed. 1968;109:504–512. - PubMed
-
- Whyte MP. Osteopetrosis. In: Royce PM, Steinmann B, editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2nd ed. Vol. 2002. New York: Wiley-Liss; pp. 789–807.
-
- Leisti J, Kaitila I, Lachman RS, Asch MJ, Rimoin DL. Dysosteosclerosis. Birth Defects. 1975;11:349–351. - PubMed
-
- Kaitila I, Rimoin DL. Histologic heterogeneity in the hyperostotic bone dysplasias. Birth Defects. 1976;21:71–79. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
