

Induction of clusterin by AKT--role in cytoprotection against docetaxel in prostate tumor cells
- PMID: 20501799
- PMCID: PMC3874868
- DOI: 10.1158/1535-7163.MCT-09-0880
Induction of clusterin by AKT--role in cytoprotection against docetaxel in prostate tumor cells
Abstract
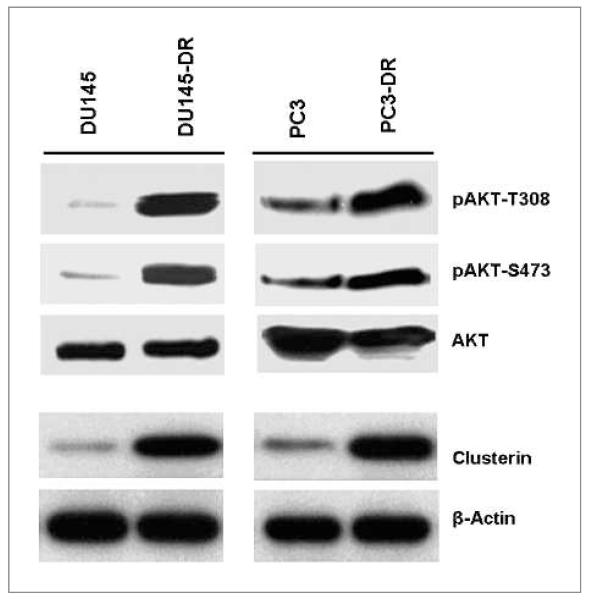
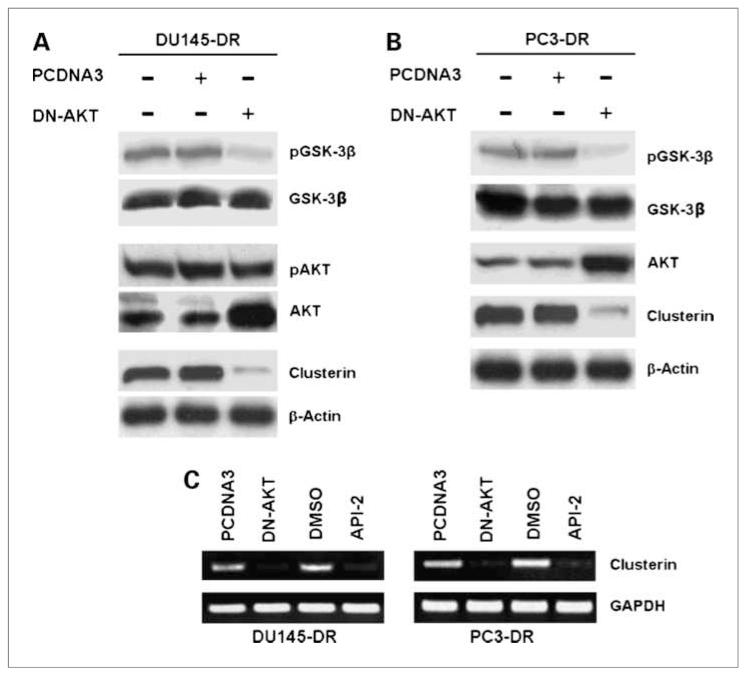
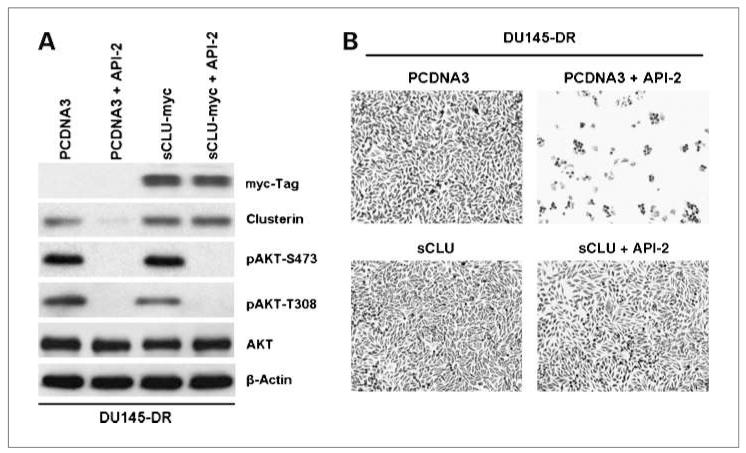
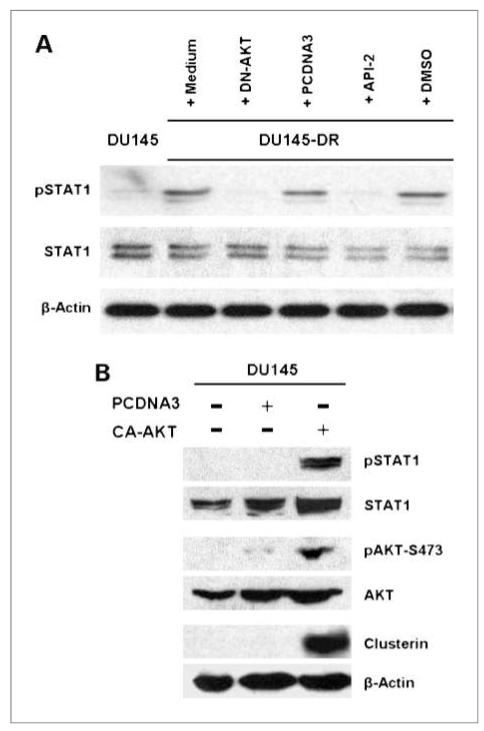
Clusterin (CLU), in its cytoplasmic form, is abundant in many advanced cancers and has been established to be cytoprotective against chemotherapeutic agents including docetaxel. However, little is known of the mechanism of its induction. Here, we provide evidence that AKT plays a critical role in upregulating cytoplasmic/secretory sCLU, which is responsible for docetaxel resistance. Western blot analysis indicated that docetaxel-resistant sublines derived from DU145 and PC3 prostate tumor cell lines displayed a markedly increased phospho-AKT level closely accompanied by heightened sCLU expression when compared with parental cells. To examine if AKT has a role in sCLU expression, AKT blockade was done by treatment with a specific inhibitor, API-2, or dominant-negative AKT transduction before analysis of sCLU gene expression. Loss of AKT function resulted in loss of sCLU and was accompanied by chemosensitization to docetaxel and increased cell death via a caspase-3-dependent pathway. To confirm that AKT affected resistance to docetaxel through sCLU and not through other mediators, tumor cells were first transfected with full-length CLU for overexpression and then treated with the AKT inhibitor API-2. We found that once sCLU was overexpressed, API-2 could not chemosensitize the tumor cells to docetaxel. Thus, the chemoresistance to docetaxel is mediated by sCLU and it can be induced by AKT. Lastly, AKT was found to mediate sCLU induction via signal transducer and activator of transcription 1 activation, which we have earlier shown to drive sCLU gene expression. These results identify a previously unrecognized pathway linking AKT to cytoprotection by sCLU in tumor cells.
Figures


References
-
- Hadaschik BA, Sowery RD, Gleave ME. Novel targets and approaches in advanced prostate cancer. Curr Opin Urol. 2007;17:182–7. - PubMed
-
- Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. - PubMed
-
- Shannan B, Seifert M, Leskov K, et al. Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006;13:12–9. - PubMed
-
- Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7:909–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
