

The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development
- PMID: 20501936
- PMCID: PMC2902546
- DOI: 10.1126/scisignal.2000727
The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development
Abstract
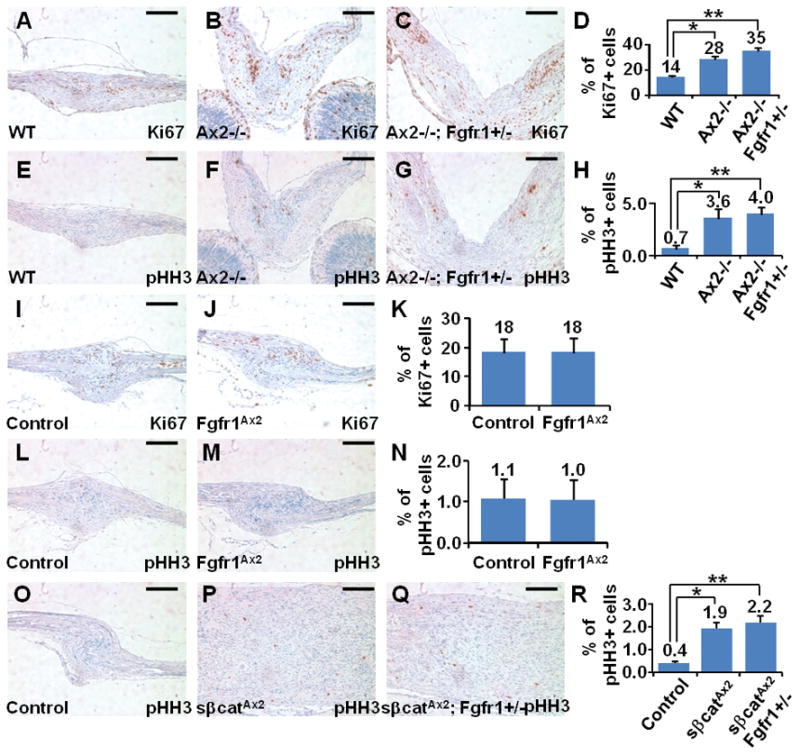
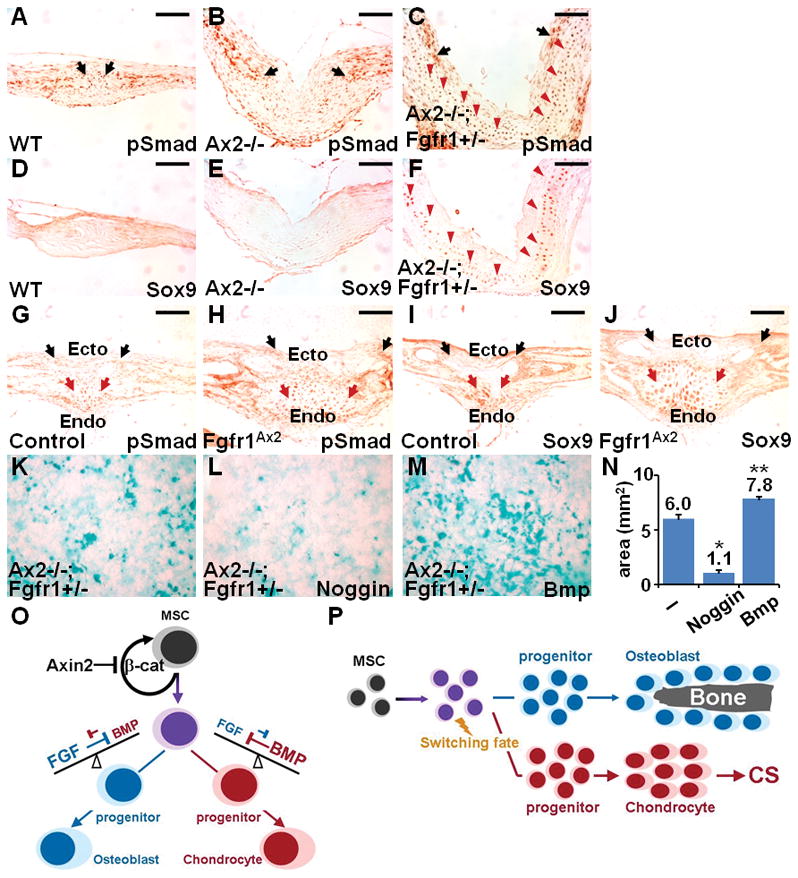
Craniosynostosis, a developmental disorder resulting from premature closure of the gaps (sutures) between skull bones, can be caused by excessive intramembranous ossification, a type of bone formation that does not involve formation of a cartilage template (chondrogenesis). Here, we show that endochondral ossification, a type of bone formation that proceeds through a cartilage intermediate, caused by switching the fate of mesenchymal stem cells to chondrocytes, can also result in craniosynostosis. Simultaneous knockout of Axin2, a negative regulator of the WNT-beta-catenin pathway, and decreased activity of fibroblast growth factor (FGF) receptor 1 (FGFR1) in mice induced ectopic chondrogenesis, leading to abnormal suture morphogenesis and fusion. Genetic analyses revealed that activation of beta-catenin cooperated with FGFR1 to alter the lineage commitment of mesenchymal stem cells to differentiate into chondrocytes, from which cartilage is formed. We showed that the WNT-beta-catenin pathway directly controlled the stem cell population by regulating its renewal and proliferation, and indirectly modulated lineage specification by setting the balance of the FGF and bone morphogenetic protein pathways. This study identifies endochondral ossification as a mechanism of suture closure during development and implicates this process in craniosynostosis.
Conflict of interest statement
Figures

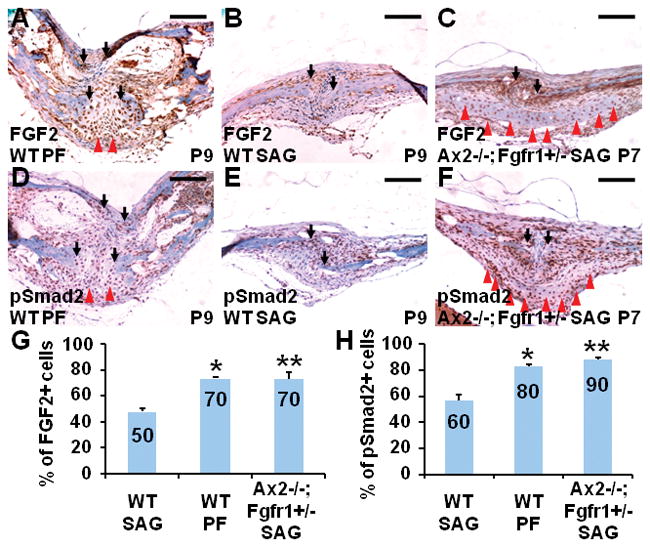
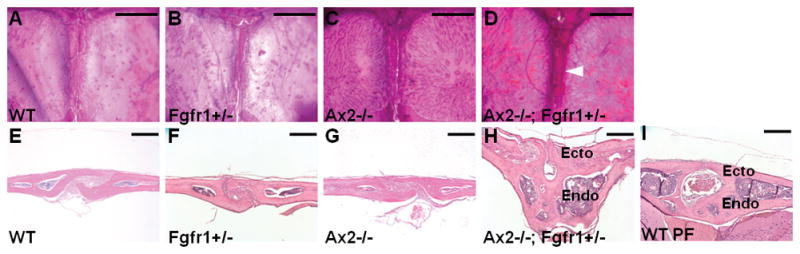
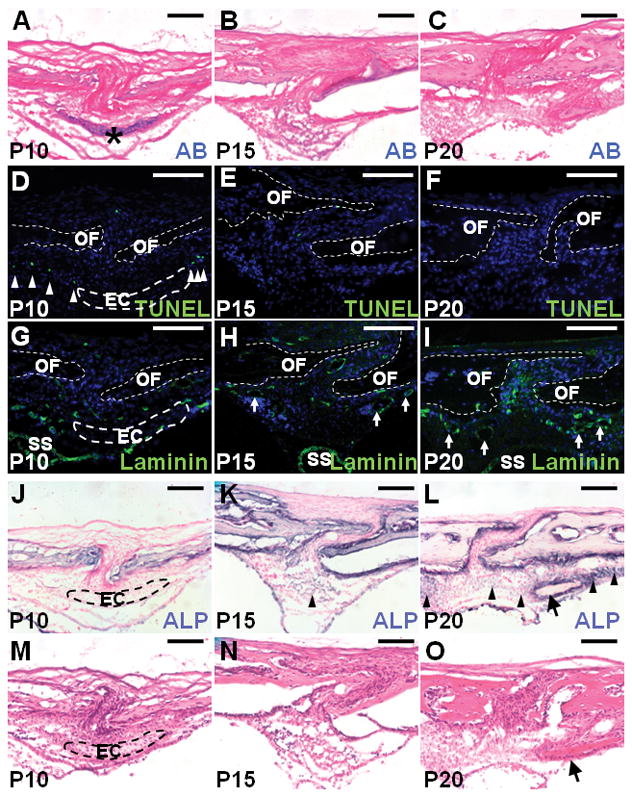
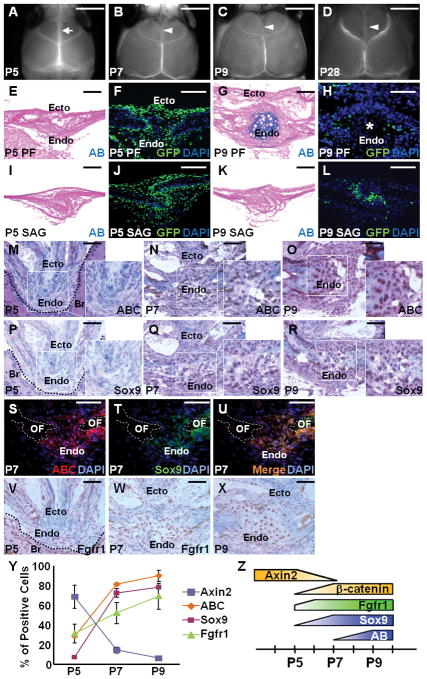


References
-
- Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2:458–468. - PubMed
-
- Cohen MM, MacLean RE. Craniosynostosis: Diagnosis, Evaluation, and Management. Oxford University Press; New York: 2000. p. 454.
-
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219:472–485. - PubMed
-
- Hall BK. Bone. Telford Press; Caldwell, NJ: 1990. p. v.
-
- Wilkie AO. Craniosynostosis: Genes and mechanisms. Hum Mol Genet. 1997;6:1647–1656. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
