

Analysis and computational dissection of molecular signature multiplicity
- PMID: 20502670
- PMCID: PMC2873900
- DOI: 10.1371/journal.pcbi.1000790
Analysis and computational dissection of molecular signature multiplicity
Abstract
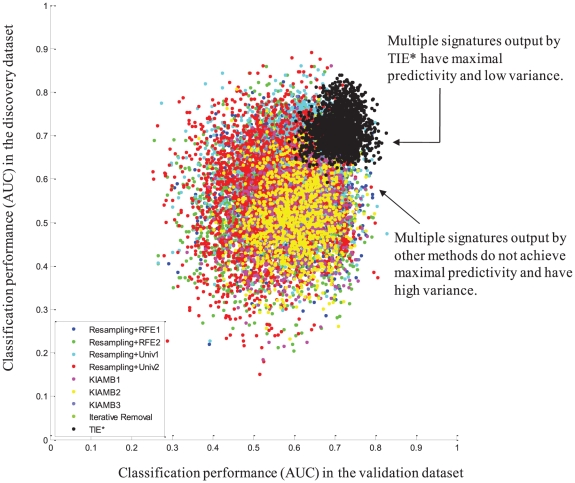
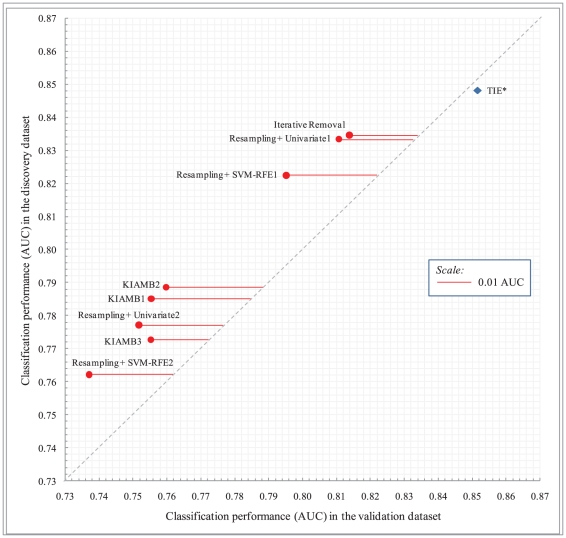
Molecular signatures are computational or mathematical models created to diagnose disease and other phenotypes and to predict clinical outcomes and response to treatment. It is widely recognized that molecular signatures constitute one of the most important translational and basic science developments enabled by recent high-throughput molecular assays. A perplexing phenomenon that characterizes high-throughput data analysis is the ubiquitous multiplicity of molecular signatures. Multiplicity is a special form of data analysis instability in which different analysis methods used on the same data, or different samples from the same population lead to different but apparently maximally predictive signatures. This phenomenon has far-reaching implications for biological discovery and development of next generation patient diagnostics and personalized treatments. Currently the causes and interpretation of signature multiplicity are unknown, and several, often contradictory, conjectures have been made to explain it. We present a formal characterization of signature multiplicity and a new efficient algorithm that offers theoretical guarantees for extracting the set of maximally predictive and non-redundant signatures independent of distribution. The new algorithm identifies exactly the set of optimal signatures in controlled experiments and yields signatures with significantly better predictivity and reproducibility than previous algorithms in human microarray gene expression datasets. Our results shed light on the causes of signature multiplicity, provide computational tools for studying it empirically and introduce a framework for in silico bioequivalence of this important new class of diagnostic and personalized medicine modalities.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Network based consensus gene signatures for biomarker discovery in breast cancer.PLoS One. 2011;6(10):e25364. doi: 10.1371/journal.pone.0025364. Epub 2011 Oct 25. PLoS One. 2011. PMID: 22046239 Free PMC article.
-
Outcome prediction based on microarray analysis: a critical perspective on methods.BMC Bioinformatics. 2009 Feb 7;10:53. doi: 10.1186/1471-2105-10-53. BMC Bioinformatics. 2009. PMID: 19200394 Free PMC article.
-
A parallel and incremental algorithm for efficient unique signature discovery on DNA databases.BMC Bioinformatics. 2010 Mar 16;11:132. doi: 10.1186/1471-2105-11-132. BMC Bioinformatics. 2010. PMID: 20230647 Free PMC article.
-
Expanding the understanding of biases in development of clinical-grade molecular signatures: a case study in acute respiratory viral infections.PLoS One. 2011;6(6):e20662. doi: 10.1371/journal.pone.0020662. Epub 2011 Jun 1. PLoS One. 2011. PMID: 21673802 Free PMC article.
-
Molecular signatures from omics data: from chaos to consensus.Biotechnol J. 2012 Aug;7(8):946-57. doi: 10.1002/biot.201100305. Epub 2012 Apr 23. Biotechnol J. 2012. PMID: 22528809 Free PMC article. Review.
Cited by
-
A biological function based biomarker panel optimization process.Sci Rep. 2019 May 14;9(1):7365. doi: 10.1038/s41598-019-43779-2. Sci Rep. 2019. PMID: 31089177 Free PMC article.
-
Extending greedy feature selection algorithms to multiple solutions.Data Min Knowl Discov. 2021;35(4):1393-1434. doi: 10.1007/s10618-020-00731-7. Epub 2021 May 1. Data Min Knowl Discov. 2021. PMID: 34720675 Free PMC article.
-
Predictive integration of gene functional similarity and co-expression defines treatment response of endothelial progenitor cells.BMC Syst Biol. 2011 Mar 30;5:46. doi: 10.1186/1752-0509-5-46. BMC Syst Biol. 2011. PMID: 21447198 Free PMC article.
-
Gene regulatory network inference using fused LASSO on multiple data sets.Sci Rep. 2016 Feb 11;6:20533. doi: 10.1038/srep20533. Sci Rep. 2016. PMID: 26864687 Free PMC article.
-
Multidimensional Integrative Genomics Approaches to Dissecting Cardiovascular Disease.Front Cardiovasc Med. 2017 Feb 27;4:8. doi: 10.3389/fcvm.2017.00008. eCollection 2017. Front Cardiovasc Med. 2017. PMID: 28289683 Free PMC article. Review.
References
-
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. - PubMed
-
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. - PubMed
-
- Azuaje F, Dopazo J. Hoboken, NJ: John Wiley; 2005. Data analysis and visualization in genomics and proteomics.
-
- Somorjai RL, Dolenko B, Baumgartner R. Class prediction and discovery using gene microarray and proteomics mass spectroscopy data: curses, caveats, cautions. Bioinformatics. 2003;19:1484–1491. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
