

Novel peptide-mediated interactions derived from high-resolution 3-dimensional structures
- PMID: 20502673
- PMCID: PMC2873903
- DOI: 10.1371/journal.pcbi.1000789
Novel peptide-mediated interactions derived from high-resolution 3-dimensional structures
Abstract
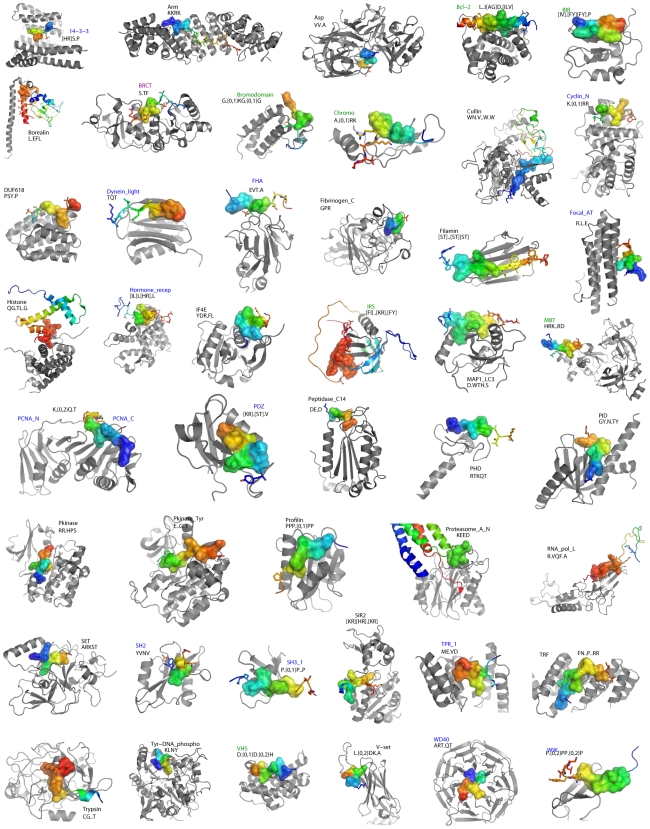
Many biological responses to intra- and extracellular stimuli are regulated through complex networks of transient protein interactions where a globular domain in one protein recognizes a linear peptide from another, creating a relatively small contact interface. These peptide stretches are often found in unstructured regions of proteins, and contain a consensus motif complementary to the interaction surface displayed by their binding partners. While most current methods for the de novo discovery of such motifs exploit their tendency to occur in disordered regions, our work here focuses on another observation: upon binding to their partner domain, motifs adopt a well-defined structure. Indeed, through the analysis of all peptide-mediated interactions of known high-resolution three-dimensional (3D) structure, we found that the structure of the peptide may be as characteristic as the consensus motif, and help identify target peptides even though they do not match the established patterns. Our analyses of the structural features of known motifs reveal that they tend to have a particular stretched and elongated structure, unlike most other peptides of the same length. Accordingly, we have implemented a strategy based on a Support Vector Machine that uses this features, along with other structure-encoded information about binding interfaces, to search the set of protein interactions of known 3D structure and to identify unnoticed peptide-mediated interactions among them. We have also derived consensus patterns for these interactions, whenever enough information was available, and compared our results with established linear motif patterns and their binding domains. Finally, to cross-validate our identification strategy, we scanned interactome networks from four model organisms with our newly derived patterns to see if any of them occurred more often than expected. Indeed, we found significant over-representations for 64 domain-motif interactions, 46 of which had not been described before, involving over 6,000 interactions in total for which we could suggest the molecular details determining the binding.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
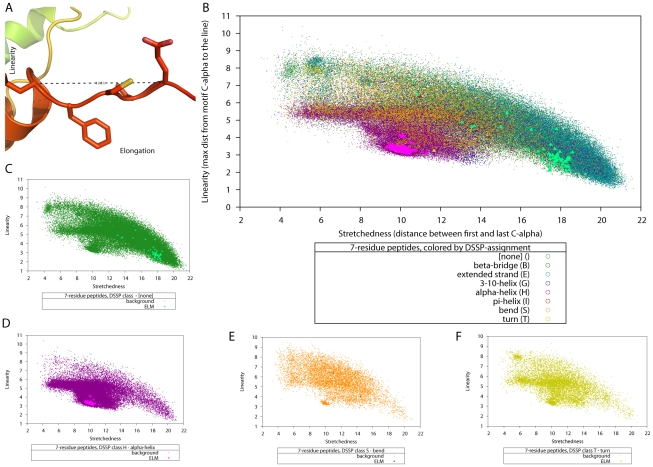
 in the motif from the line through the first and last
in the motif from the line through the first and last  ) and elongation (the distance between the first and last
) and elongation (the distance between the first and last  of a motif) are illustrated in this structure. (B) A slice of the data used for SVM training: linearity, elongation and secondary structure classification for 7-residue-peptides, with data from the SCOP background shown as dots and the data for known DMI shown as solid triangles, using one colour per DSSP classification. Panels (C) to (F) show the distribution of linearity∶elongation values for those secondary structure classifications for which we had known 7-residue-peptides (none, alpha-helix, bend, and turn). These data slices illustrate how known linear motifs fall into distinct regions of the parameter space.
of a motif) are illustrated in this structure. (B) A slice of the data used for SVM training: linearity, elongation and secondary structure classification for 7-residue-peptides, with data from the SCOP background shown as dots and the data for known DMI shown as solid triangles, using one colour per DSSP classification. Panels (C) to (F) show the distribution of linearity∶elongation values for those secondary structure classifications for which we had known 7-residue-peptides (none, alpha-helix, bend, and turn). These data slices illustrate how known linear motifs fall into distinct regions of the parameter space.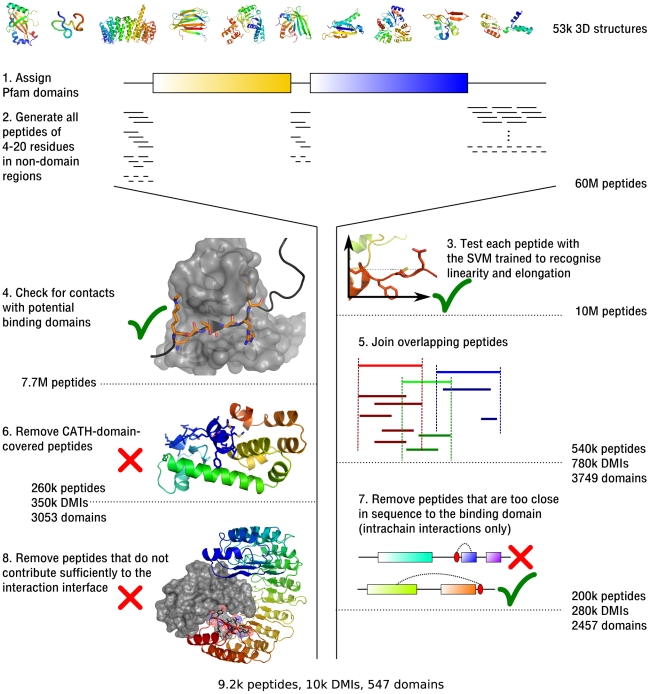
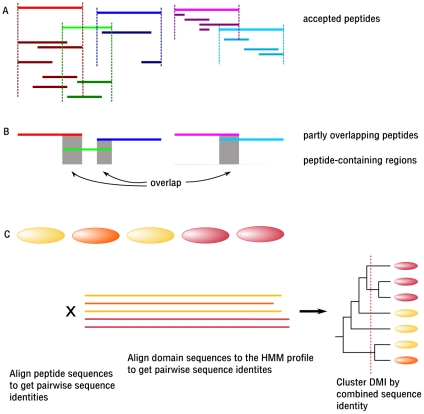
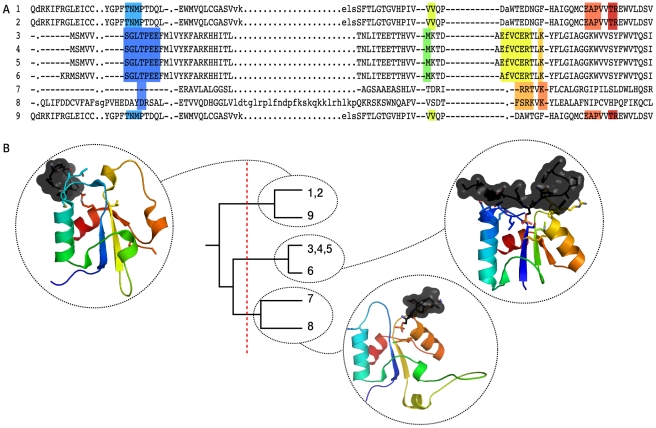
References
-
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. - PubMed
-
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. - PubMed
-
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. - PubMed
-
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
