

Co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy
- PMID: 20503315
- PMCID: PMC4048012
- DOI: 10.1002/ajmg.a.33416
Co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy
Abstract
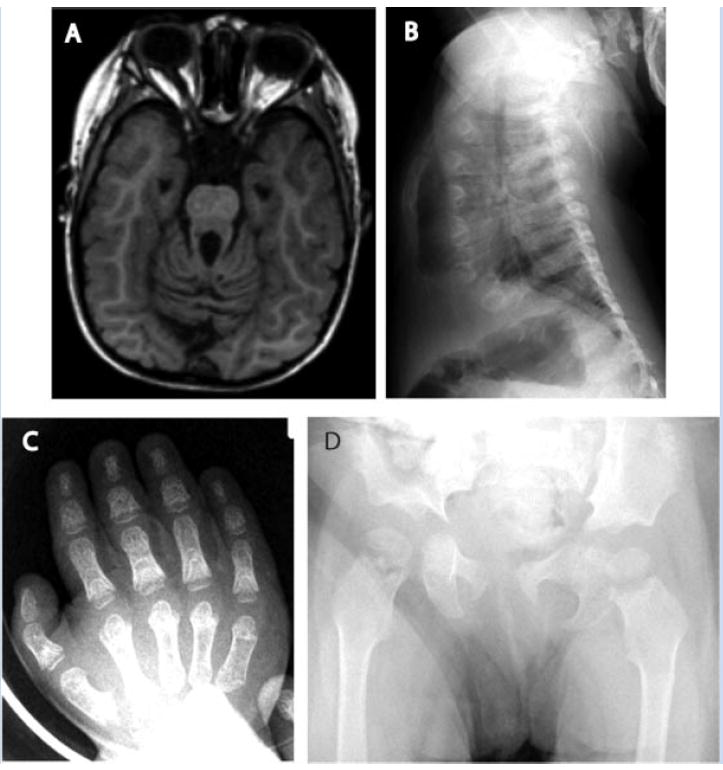
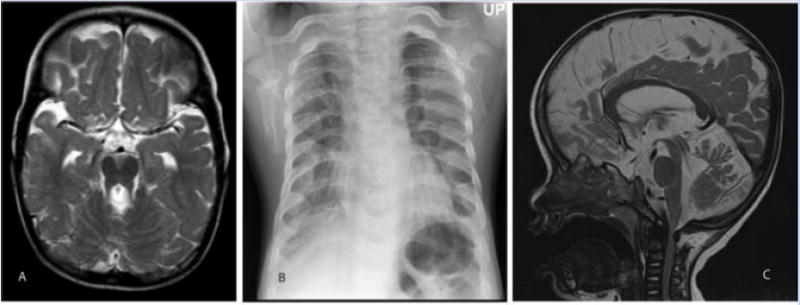
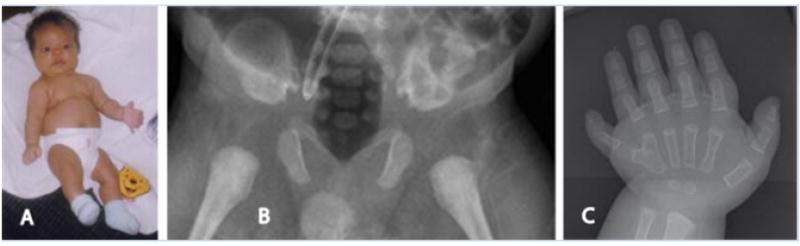
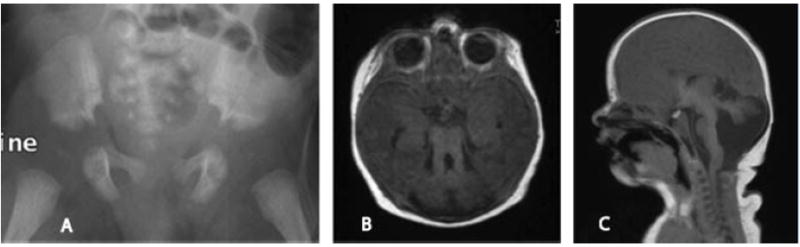
Ciliary disorders share typical features, such as polydactyly, renal and biliary cystic dysplasia, and retinitis pigmentosa, which often overlap across diagnostic entities. We report on two siblings of consanguineous parents and two unrelated children, both of unrelated parents, with co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy, an association that adds to the observation of common final patterns of malformations in ciliary disorders. Using homozygosity mapping in the siblings, we were able to exclude all known genes/loci for both syndromes except for INVS, AHI1, and three genes from the previously described Jeune locus at 15q13. No pathogenic variants were found in these genes by direct sequencing. In the third child reported, sequencing of RPGRIP1L, ARL13B, AHI1, TMEM67, OFD1, CC2D2A, and deletion analysis of NPHP1 showed no mutations. Although this study failed to identify a mutation in the patients tested, the co-occurrence of Joubert and Jeune syndromes is likely to represent a distinct entity caused by mutations in a yet to be discovered gene. The mechanisms by which certain organ systems are affected more than others in the spectrum of ciliary diseases remain largely unknown.
(c) 2010 Wiley-Liss, Inc.
Figures
Similar articles
-
Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes.J Med Genet. 2015 Dec;52(12):830-9. doi: 10.1136/jmedgenet-2015-103316. Epub 2015 Sep 18. J Med Genet. 2015. PMID: 26386044 Free PMC article.
-
CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290.Am J Hum Genet. 2008 Nov;83(5):559-71. doi: 10.1016/j.ajhg.2008.10.002. Epub 2008 Oct 23. Am J Hum Genet. 2008. PMID: 18950740 Free PMC article.
-
A locus for asphyxiating thoracic dystrophy, ATD, maps to chromosome 15q13.J Med Genet. 2003 Jun;40(6):431-5. doi: 10.1136/jmg.40.6.431. J Med Genet. 2003. PMID: 12807964 Free PMC article.
-
Hepatic manifestations of Jeune syndrome (asphyxiating thoracic dystrophy).Semin Liver Dis. 2003 May;23(2):195-200. doi: 10.1055/s-2003-39950. Semin Liver Dis. 2003. PMID: 12800072 Review.
-
Clinical and molecular features of Joubert syndrome and related disorders.Am J Med Genet C Semin Med Genet. 2009 Nov 15;151C(4):326-40. doi: 10.1002/ajmg.c.30229. Am J Med Genet C Semin Med Genet. 2009. PMID: 19876931 Free PMC article. Review.
Cited by
-
Olfactory Loss and Dysfunction in Ciliopathies: Molecular Mechanisms and Potential Therapies.Curr Med Chem. 2019;26(17):3103-3119. doi: 10.2174/0929867325666180105102447. Curr Med Chem. 2019. PMID: 29303074 Free PMC article. Review.
-
A human ciliopathy with polycystic ovarian syndrome and multiple subcutaneous cysts: A rare case report.Medicine (Baltimore). 2018 Dec;97(50):e13531. doi: 10.1097/MD.0000000000013531. Medicine (Baltimore). 2018. PMID: 30558011 Free PMC article.
-
Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes.J Med Genet. 2015 Dec;52(12):830-9. doi: 10.1136/jmedgenet-2015-103316. Epub 2015 Sep 18. J Med Genet. 2015. PMID: 26386044 Free PMC article.
-
Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes.J Med Genet. 2016 Sep;53(9):608-15. doi: 10.1136/jmedgenet-2016-103832. Epub 2016 May 6. J Med Genet. 2016. PMID: 27208211 Free PMC article.
-
Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans.Am J Hum Genet. 2013 Nov 7;93(5):915-25. doi: 10.1016/j.ajhg.2013.09.012. Epub 2013 Oct 17. Am J Hum Genet. 2013. PMID: 24140113 Free PMC article.
References
-
- Ardura Fernández J, Alvarez González C, Rodríguez Fernández M, Andrés de Llano J. Asphyxiating thoracic dysplasia associated with proximal myopathy and arachnoid cyst. An Esp Pediatr. 1990;33:592–596. - PubMed
-
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. - PubMed
-
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. - PMC - PubMed
-
- Barnes EG, Opitz JM. Renal abnormalities in malformation syndromes. In: Edelmann CM Jr, Bernstein J, Meadow SR, Spitzer A, Travis LB, editors. Pediatric kidney disease. 2. Boston: Little Brown; 1992. pp. 1067–1119.
-
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tüysüz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007;39:727–729. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
