

Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome 1
- PMID: 20503323
- PMCID: PMC2947940
- DOI: 10.1002/ajmg.a.33389
Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome 1
Abstract
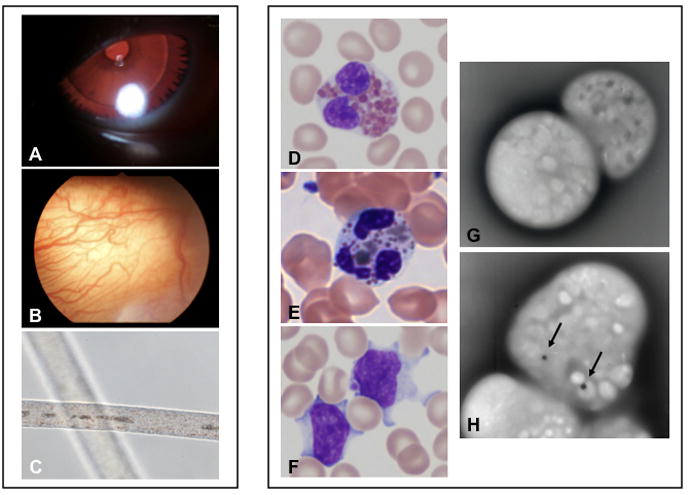
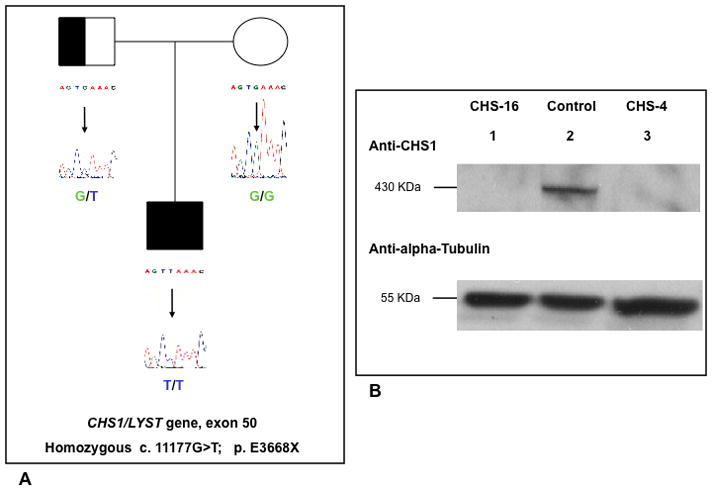
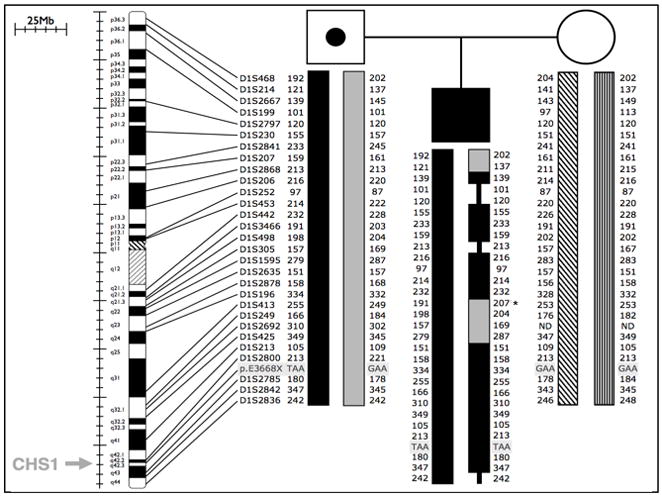
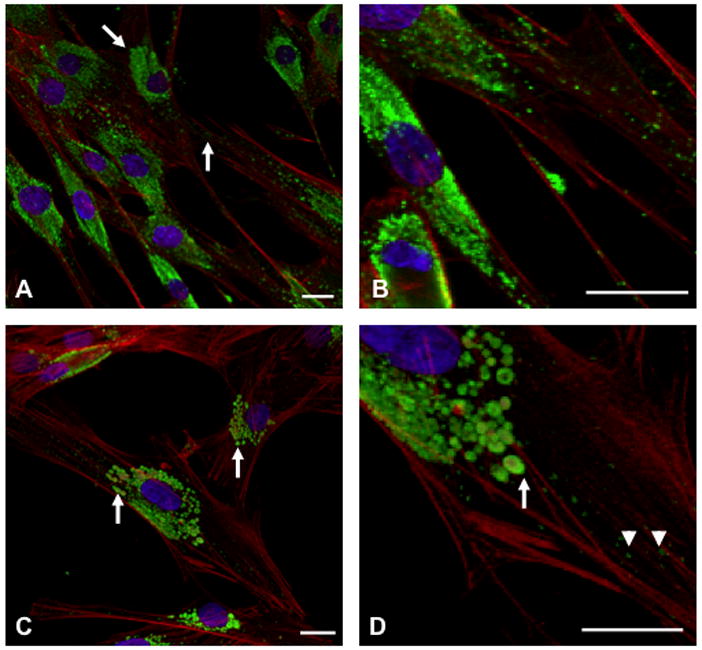
Chediak-Higashi syndrome (CHS) is a rare autosomal recessive disease characterized by variable oculocutaneous albinism, immunodeficiency, mild bleeding diathesis, and an accelerated lymphoproliferative state. Abnormal lysosome-related organelle membrane function leads to the accumulation of large intracellular vesicles in several cell types, including granulocytes, melanocytes, and platelets. This report describes a severe case of CHS resulting from paternal heterodisomy of chromosome 1, causing homozygosity for the most distal nonsense mutation (p.E3668X, exon 50) reported to date in the LYST/CHS1 gene. The mutation is located in the WD40 region of the CHS1 protein. The patient's fibroblasts expressed no detectable CHS1. Besides manifesting the classical CHS findings, the patient exhibited hypotonia and global developmental delays, raising concerns about other effects of heterodisomy. An interstitial 747 kb duplication on 6q14.2-6q14.3 was identified in the propositus and paternal samples by comparative genomic hybridization. SNP genotyping revealed no additional whole chromosome or segmental isodisomic regions or other dosage variations near the crossover breakpoints on chromosome 1. Unmasking of a separate autosomal recessive cause of developmental delay, or an additive effect of the paternal heterodisomy, could underlie the severity of the phenotype in this patient.
Published 2010 Wiley-Liss, Inc.
Figures
References
-
- Altug-Teber O, Dufke A, Poths S, Mau-Holzmann UA, Bastepe M, Colleaux L, Cormier-Daire V, Eggermann T, Gillessen-Kaesbach G, Bonin M, Riess O. A rapid microarray based whole genome analysis for detection of uniparental disomy. Hum Mutat. 2005;26:153–159. - PubMed
-
- Andersen CL, Wiuf C, Kruhoffer M, Korsgaard M, Laurberg S, Orntoft TF. Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis. 2007;28:38–48. - PubMed
-
- Boland E, Clayton-Smith J, Woo VG, McKee S, Manson FD, Medne L, Zackai E, Swanson EA, Fitzpatrick D, Millen KJ, Sherr EH, Dobyns WB, Black GC. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet. 2007;81:292–303. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
