

A comparison of probe-level and probeset models for small-sample gene expression data
- PMID: 20504334
- PMCID: PMC2901368
- DOI: 10.1186/1471-2105-11-281
A comparison of probe-level and probeset models for small-sample gene expression data
Abstract
Background: Statistical methods to tentatively identify differentially expressed genes in microarray studies typically assume larger sample sizes than are practical or even possible in some settings.
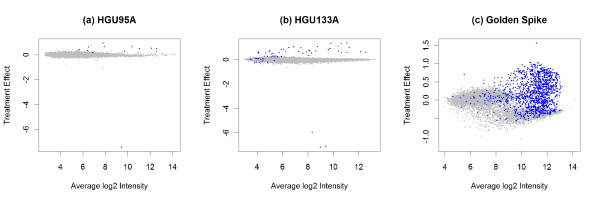
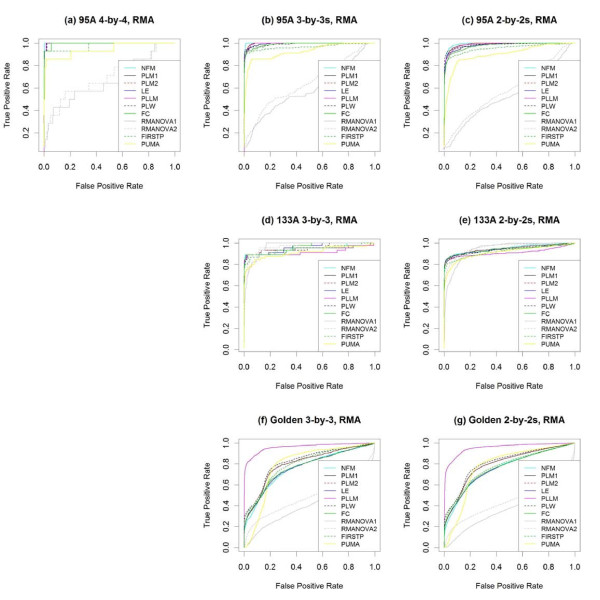
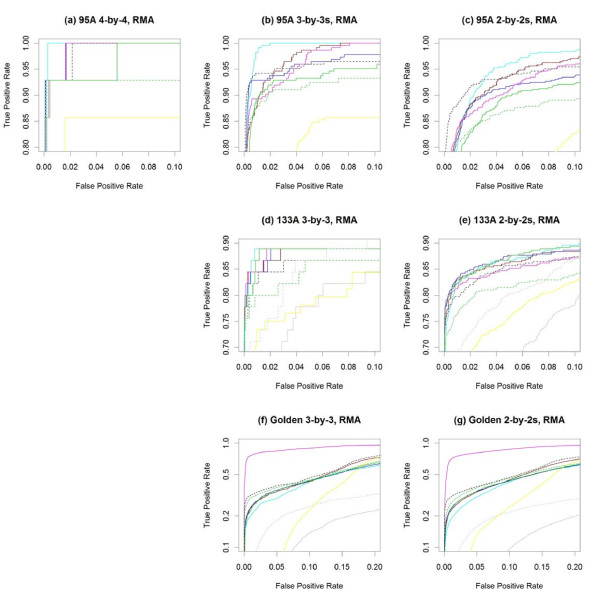
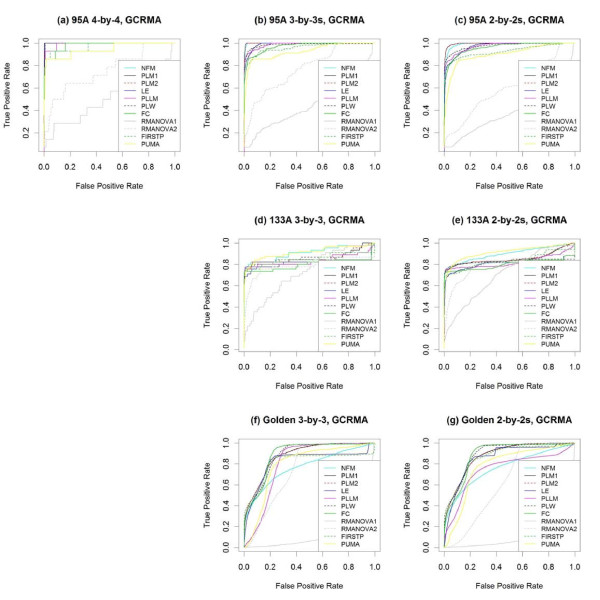
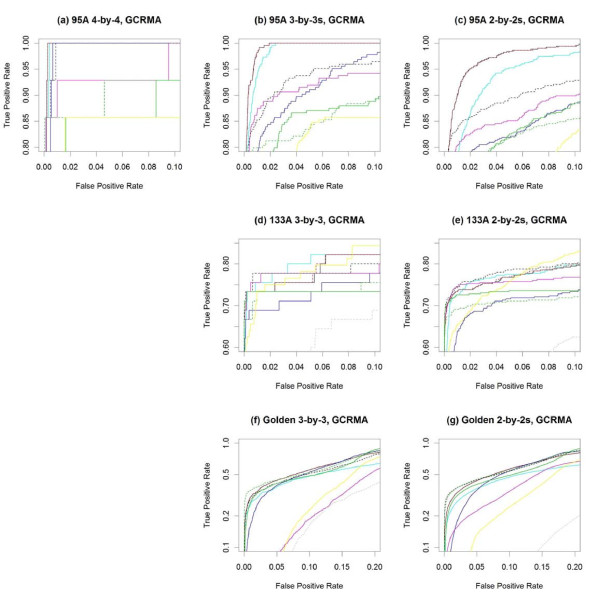
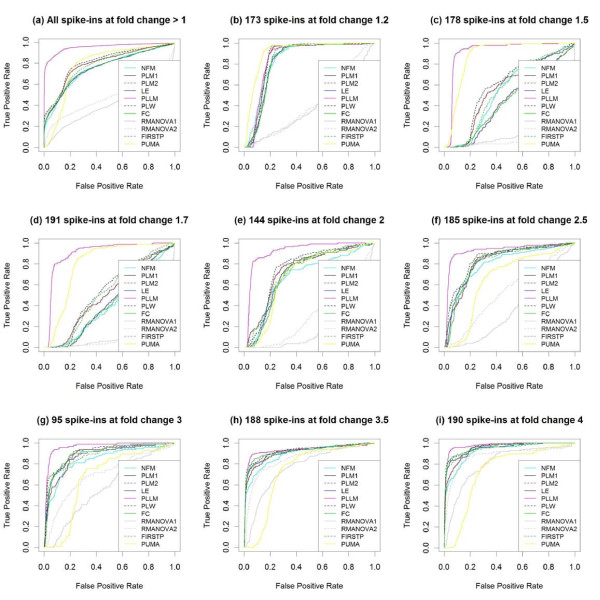
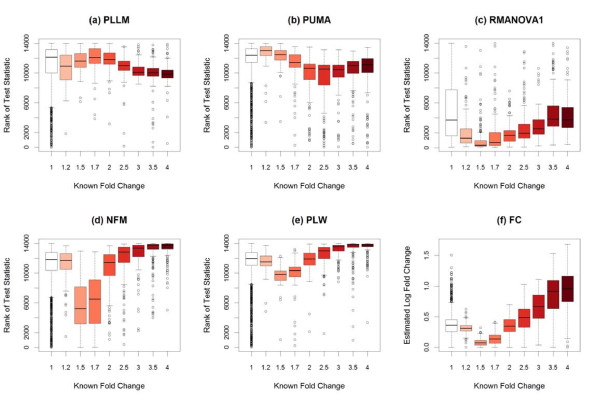
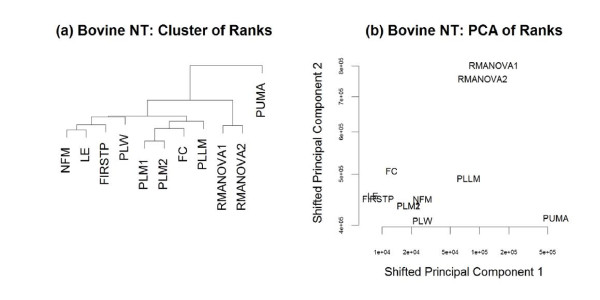
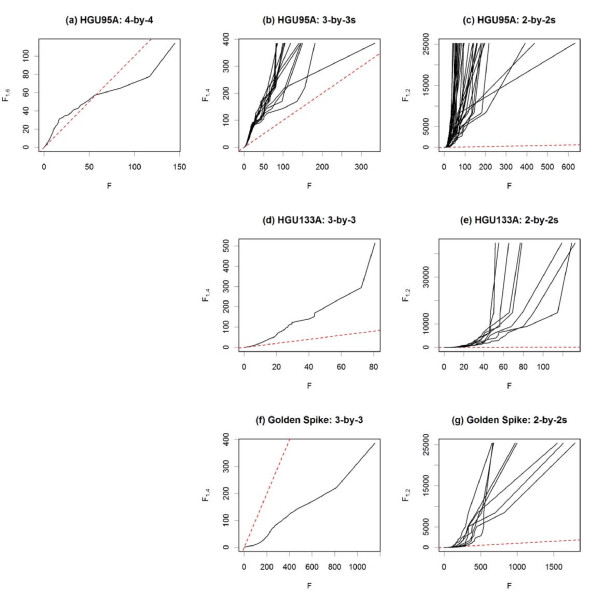
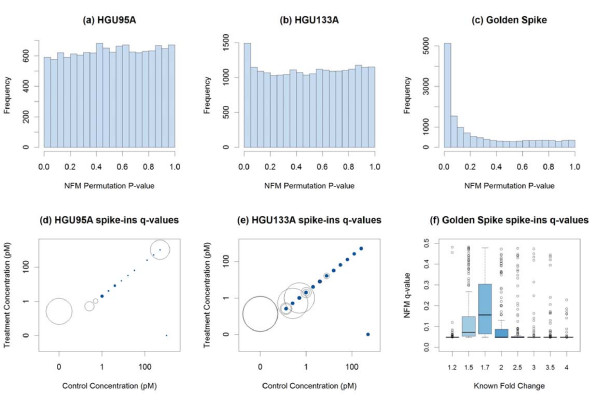
Results: The performance of several probe-level and probeset models was assessed graphically and numerically using three spike-in datasets. Based on the Affymetrix GeneChip, a novel nested factorial model was developed and found to perform competitively on small-sample spike-in experiments.
Conclusions: Statistical methods with test statistics related to the estimated log fold change tend to be more consistent in their performance on small-sample gene expression data. For such small-sample experiments, the nested factorial model can be a useful statistical tool. This method is implemented in freely-available R code (affyNFM), available with a tutorial document at http://www.stat.usu.edu/~jrstevens.
Figures
Similar articles
-
Interactively optimizing signal-to-noise ratios in expression profiling: project-specific algorithm selection and detection p-value weighting in Affymetrix microarrays.Bioinformatics. 2004 Nov 1;20(16):2534-44. doi: 10.1093/bioinformatics/bth280. Epub 2004 Apr 29. Bioinformatics. 2004. PMID: 15117752
-
Statistical analysis of high-density oligonucleotide arrays: a multiplicative noise model.Bioinformatics. 2002 Dec;18(12):1633-40. doi: 10.1093/bioinformatics/18.12.1633. Bioinformatics. 2002. PMID: 12490448
-
Practical FDR-based sample size calculations in microarray experiments.Bioinformatics. 2005 Aug 1;21(15):3264-72. doi: 10.1093/bioinformatics/bti519. Epub 2005 Jun 2. Bioinformatics. 2005. PMID: 15932903
-
A generalized likelihood ratio test to identify differentially expressed genes from microarray data.Bioinformatics. 2004 Jan 1;20(1):100-4. doi: 10.1093/bioinformatics/btg384. Bioinformatics. 2004. PMID: 14693815
-
Using ANOVA to analyze microarray data.Biotechniques. 2004 Aug;37(2):173-5, 177. doi: 10.2144/04372TE01. Biotechniques. 2004. PMID: 15335204 Review.
Cited by
-
Ocular fibroblast types differ in their mRNA profiles--implications for fibrosis prevention after aqueous shunt implantation.Mol Vis. 2013 Jun 12;19:1321-31. Print 2013. Mol Vis. 2013. PMID: 23805039 Free PMC article.
-
t-Test at the Probe Level: An Alternative Method to Identify Statistically Significant Genes for Microarray Data.Microarrays (Basel). 2014 Dec 16;3(4):340-51. doi: 10.3390/microarrays3040340. Microarrays (Basel). 2014. PMID: 27600352 Free PMC article.
-
Assessing numerical dependence in gene expression summaries with the jackknife expression difference.PLoS One. 2012;7(8):e39570. doi: 10.1371/journal.pone.0039570. Epub 2012 Aug 2. PLoS One. 2012. PMID: 22876276 Free PMC article.
-
Accounting for dependence induced by weighted KNN imputation in paired samples, motivated by a colorectal cancer study.PLoS One. 2015 Apr 7;10(4):e0119876. doi: 10.1371/journal.pone.0119876. eCollection 2015. PLoS One. 2015. PMID: 25849489 Free PMC article.
-
Incorporation of subject-level covariates in quantile normalization of miRNA data.BMC Genomics. 2015 Dec 9;16:1045. doi: 10.1186/s12864-015-2199-4. BMC Genomics. 2015. PMID: 26653287 Free PMC article.
References
-
- Aston KI, Li GP, Hicks BA, Sessions BR, Pate BJ, Hammon DS, Bunch TD, White KL. Effect of the time interval between fusion and activation on nuclear state and development in vitro and in vivo of bovine somatic cell nuclear transfer embryos. Reproduction. 2006;131:45–51. doi: 10.1530/rep.1.00714. - DOI - PubMed
-
- Gentleman R, Huber W, Carey VJ, Irizarry RA, Dudoit S. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York, Springer; 2005.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
