

Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex
- PMID: 20505090
- PMCID: PMC2905527
- DOI: 10.1523/JNEUROSCI.0736-10.2010
Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex
Abstract
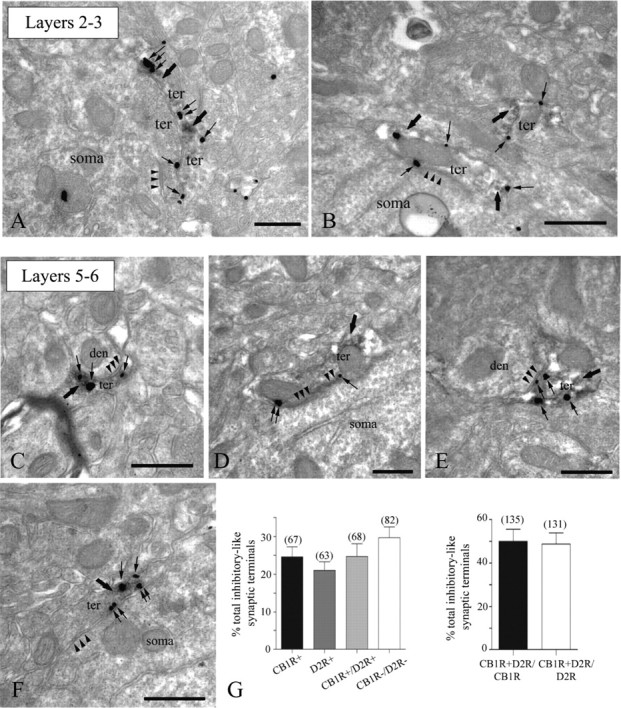
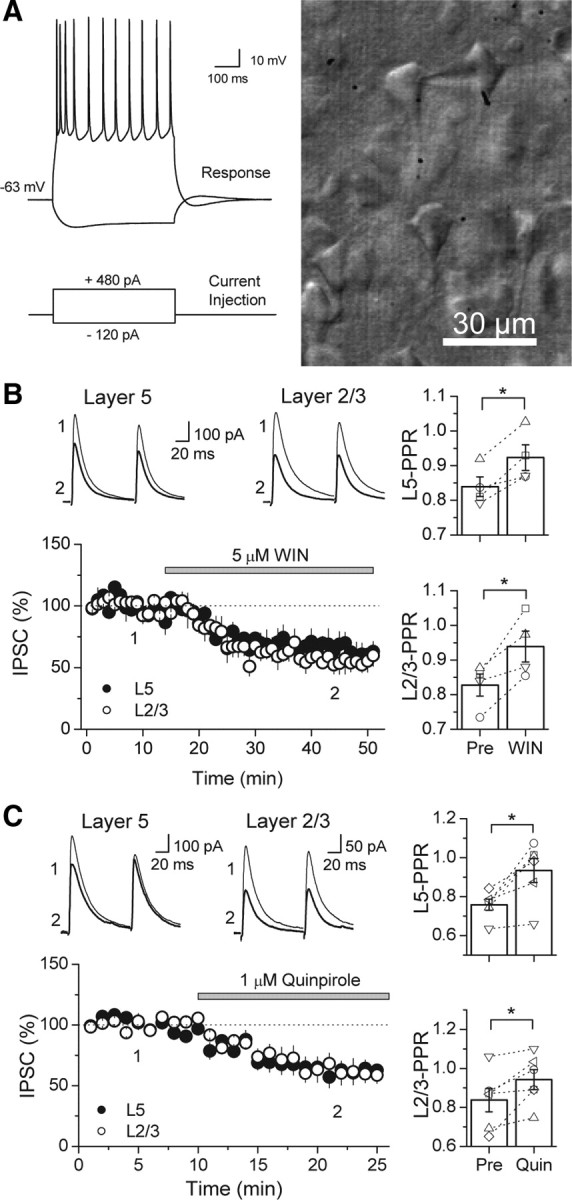
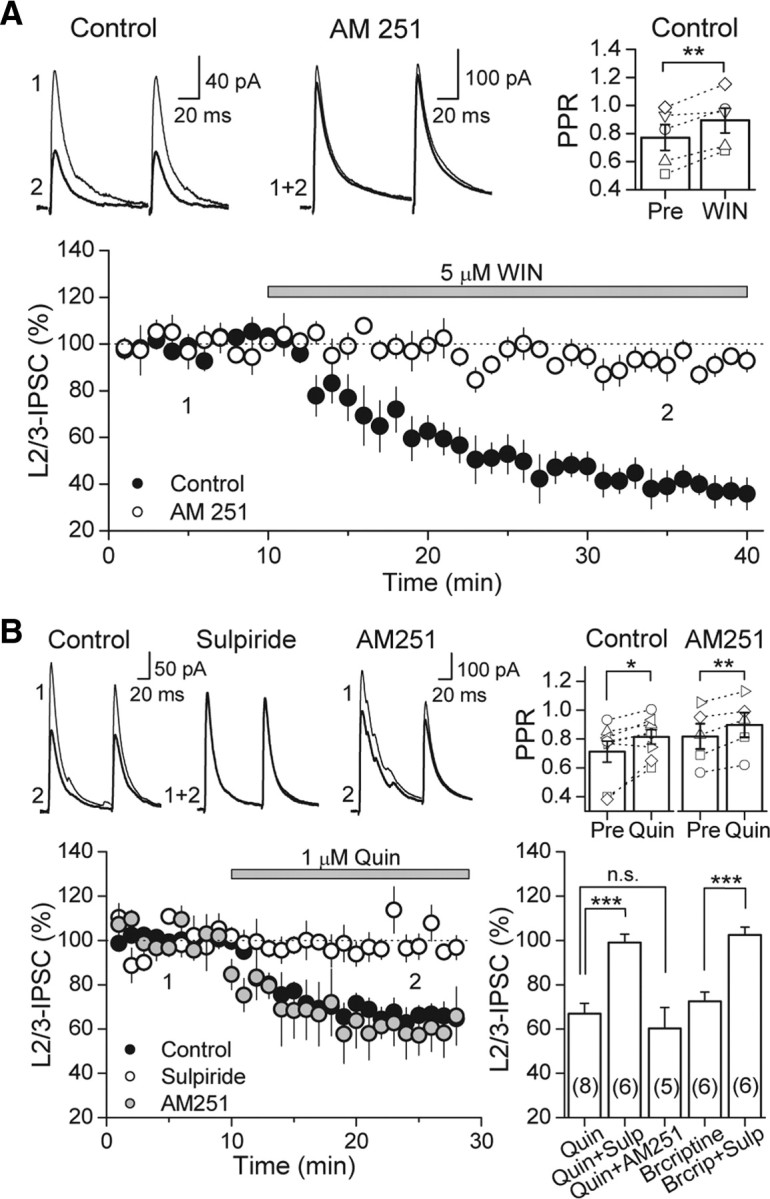
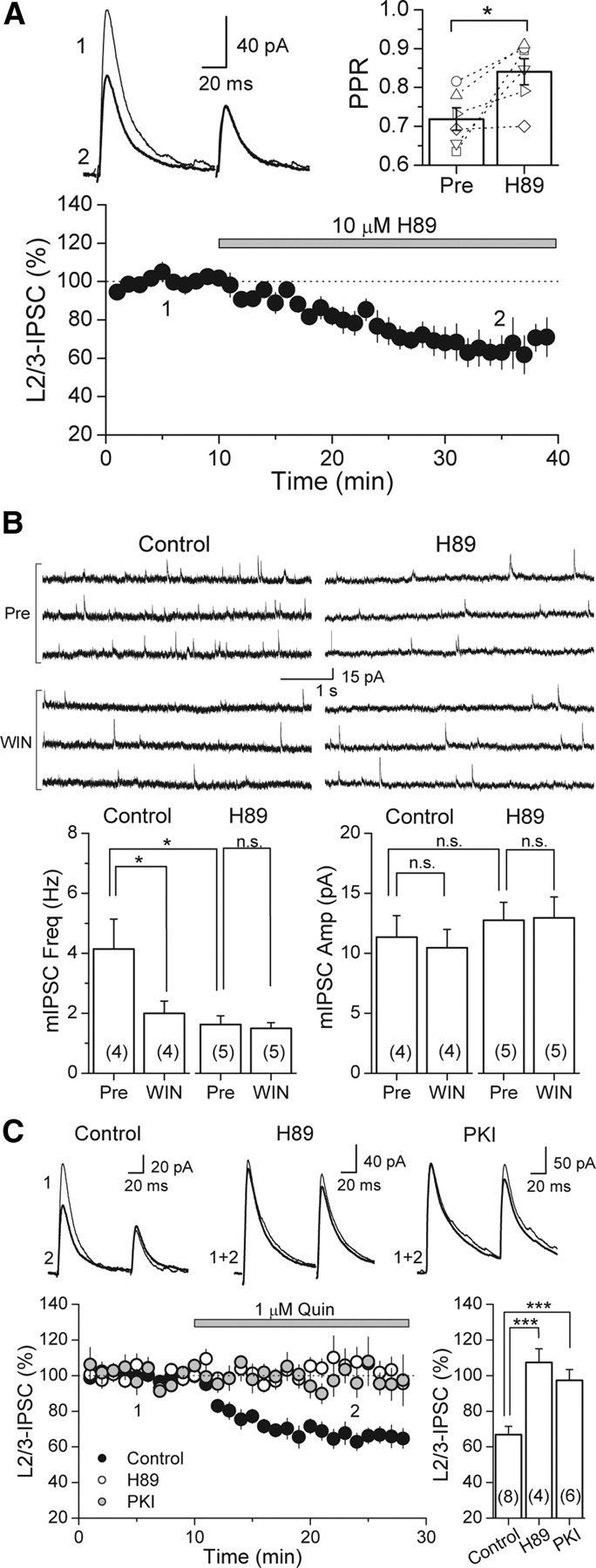
Similar to dopamine (DA), cannabinoids strongly influence prefrontal cortical functions, such as working memory, emotional learning, and sensory perception. Although endogenous cannabinoid receptors (CB(1)Rs) are abundantly expressed in the prefrontal cortex (PFC), very little is known about endocannabinoid (eCB) signaling in this brain region. Recent behavioral and electrophysiological evidence has suggested a functional interplay between the dopamine and cannabinoid receptor systems, although the cellular mechanisms underlying this interaction remain to be elucidated. We examined this issue by combining neuroanatomical and electrophysiological techniques in PFC of rats and mice (both genders). Using immunoelectron microscopy, we show that CB(1)Rs and dopamine type 2 receptors (D(2)Rs) colocalize at terminals of symmetrical, presumably GABAergic, synapses in the PFC. Indeed, activation of either receptor can suppress GABA release onto layer 5 pyramidal cells. Furthermore, coactivation of both receptors via repetitive afferent stimulation triggers eCB-mediated long-term depression of inhibitory transmission (I-LTD). This I-LTD is heterosynaptic in nature, requiring glutamate release to activate group I metabotropic glutamate receptors. D(2)Rs most likely facilitate eCB signaling at the presynaptic site as disrupting postsynaptic D(2)R signaling does not diminish I-LTD. Facilitation of eCB-LTD may be one mechanism by which DA modulates neuronal activity in the PFC and regulates PFC-mediated behavior in vivo.
Figures
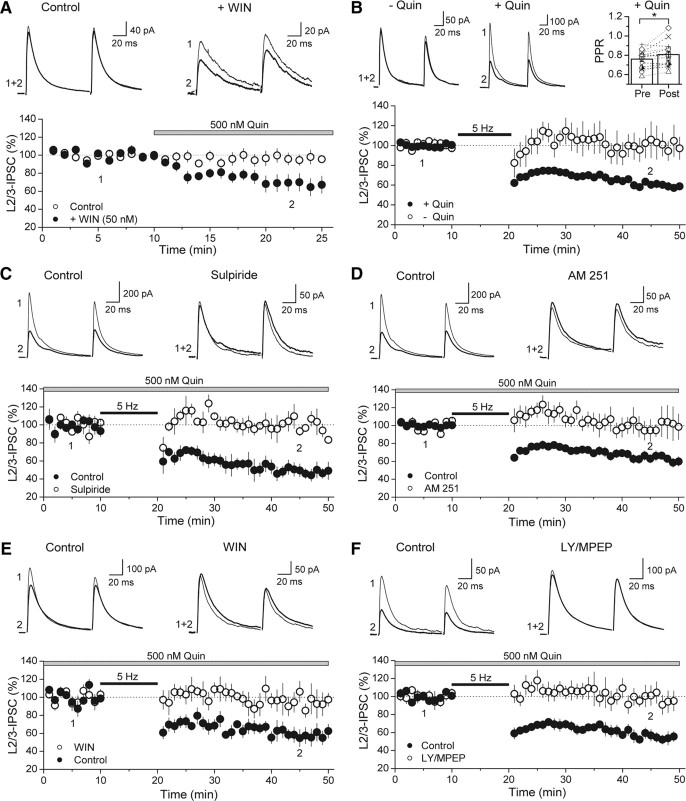
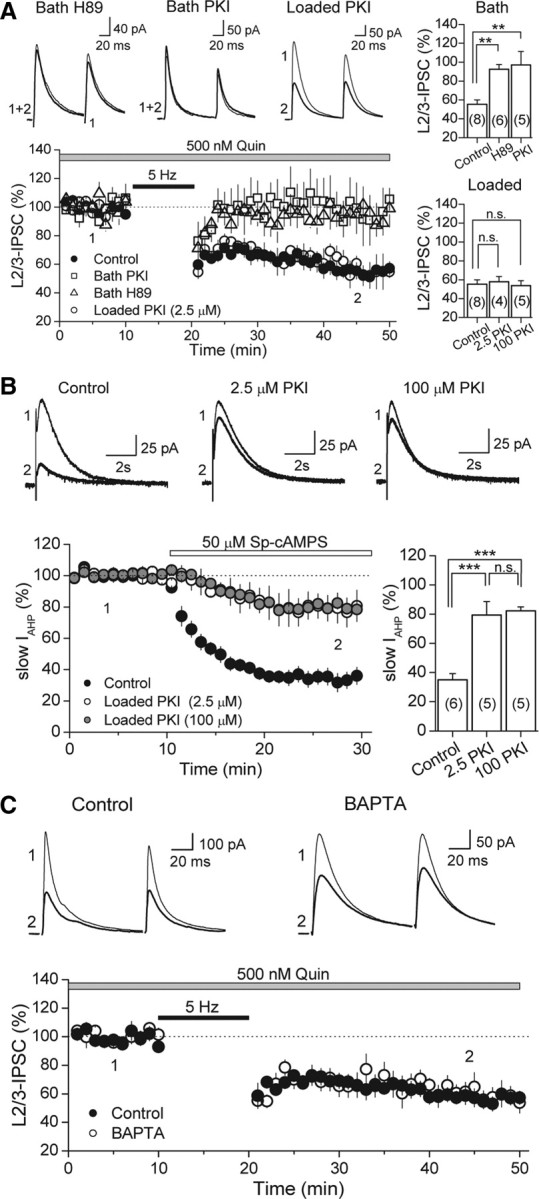
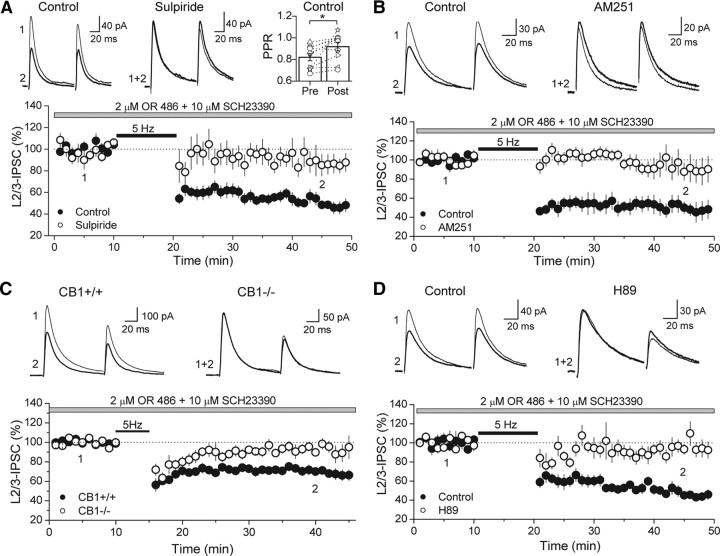
References
-
- Ariano MA, Sibley DR. Dopamine receptor distribution in the rat CNS: elucidation using anti-peptide antisera directed against D1A and D3 subtypes. Brain Res. 1994;649:95–110. - PubMed
-
- Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol. 2000;83:3287–3293. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous