

UBIAD1 mutation alters a mitochondrial prenyltransferase to cause Schnyder corneal dystrophy
- PMID: 20505825
- PMCID: PMC2874009
- DOI: 10.1371/journal.pone.0010760
UBIAD1 mutation alters a mitochondrial prenyltransferase to cause Schnyder corneal dystrophy
Abstract
Background: Mutations in a novel gene, UBIAD1, were recently found to cause the autosomal dominant eye disease Schnyder corneal dystrophy (SCD). SCD is characterized by an abnormal deposition of cholesterol and phospholipids in the cornea resulting in progressive corneal opacification and visual loss. We characterized lesions in the UBIAD1 gene in new SCD families and examined protein homology, localization, and structure.
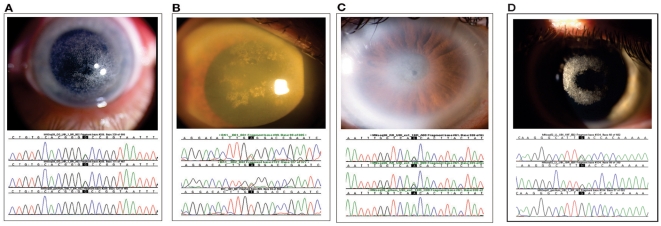
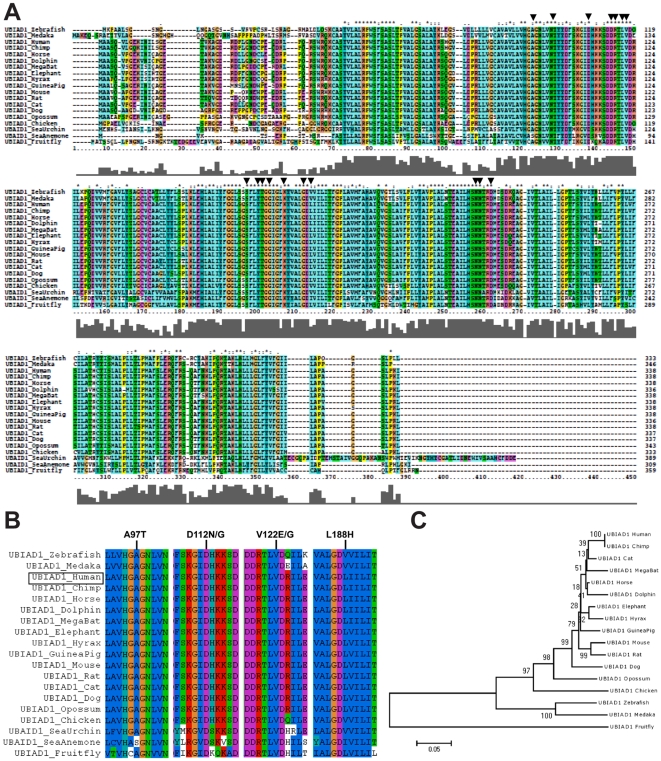
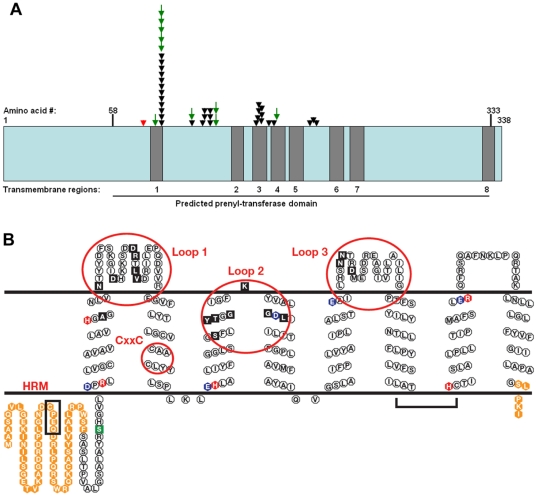
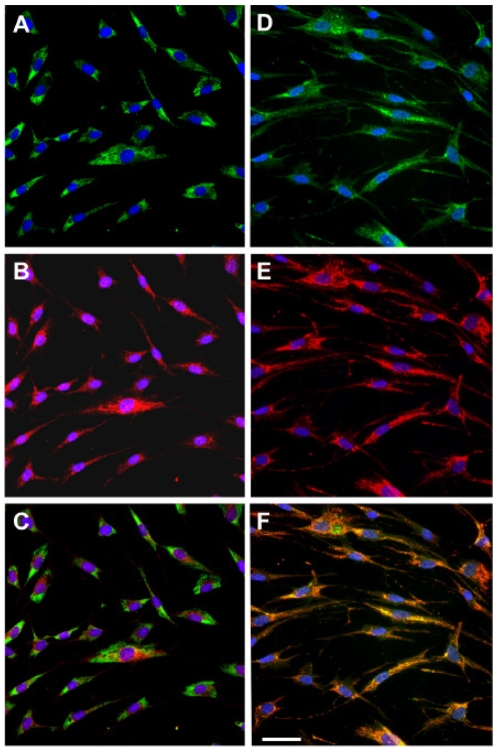
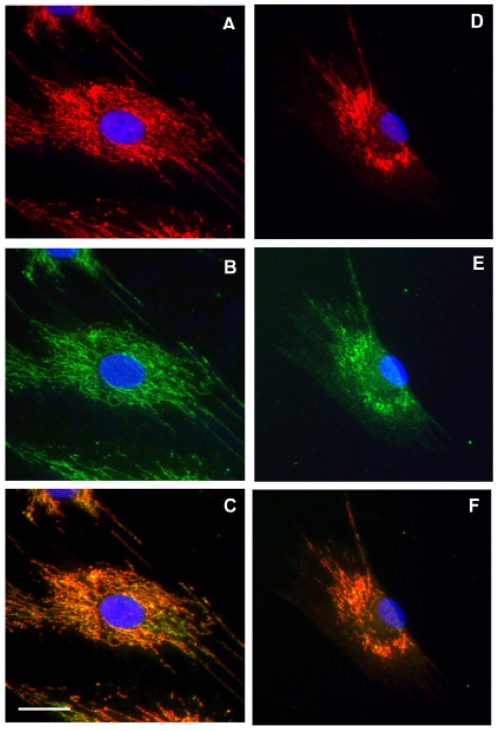
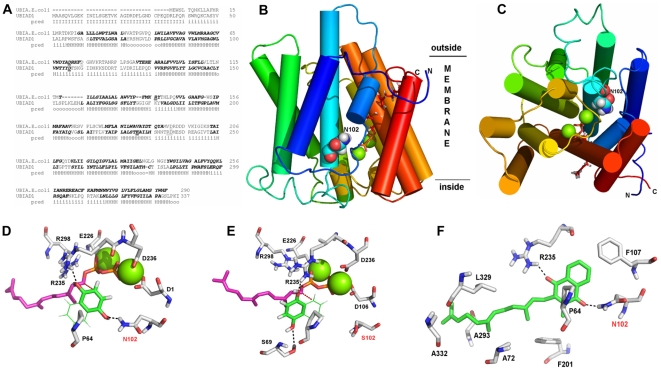
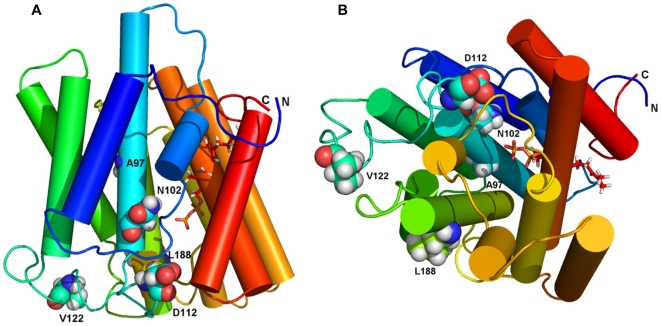
Methodology/principal findings: We characterized five novel mutations in the UBIAD1 gene in ten SCD families, including a first SCD family of Native American ethnicity. Examination of protein homology revealed that SCD altered amino acids which were highly conserved across species. Cell lines were established from patients including keratocytes obtained after corneal transplant surgery and lymphoblastoid cell lines from Epstein-Barr virus immortalized peripheral blood mononuclear cells. These were used to determine the subcellular localization of mutant and wild type protein, and to examine cholesterol metabolite ratios. Immunohistochemistry using antibodies specific for UBIAD1 protein in keratocytes revealed that both wild type and N102S protein were localized sub-cellularly to mitochondria. Analysis of cholesterol metabolites in patient cell line extracts showed no significant alteration in the presence of mutant protein indicating a potentially novel function of the UBIAD1 protein in cholesterol biochemistry. Molecular modeling was used to develop a model of human UBIAD1 protein in a membrane and revealed potentially critical roles for amino acids mutated in SCD. Potential primary and secondary substrate binding sites were identified and docking simulations indicated likely substrates including prenyl and phenolic molecules.
Conclusions/significance: Accumulating evidence from the SCD familial mutation spectrum, protein homology across species, and molecular modeling suggest that protein function is likely down-regulated by SCD mutations. Mitochondrial UBIAD1 protein appears to have a highly conserved function that, at least in humans, is involved in cholesterol metabolism in a novel manner.
Conflict of interest statement
Figures
References
-
- Van Went JM, Wibaut F. En Zeldzame erfelijk hoornvliessandoening. Niederl Tijdschr Geneesks. 1924;68:2996–2997.
-
- Schnyder WF. Mitteilung uber einen neuen typus von familiarer krankung. Schweiz Med Wschr. 1929;10:559–571.
-
- Rodrigues MM, Kruth H, Krachmer JH, Willis R. Unesterified cholesterol in Schnyder's corneal crystalline dystrophy. Am J Ophthalmol. 1987;104:157–163. - PubMed
-
- Bron AJ. Corneal changes in the dislipoproteinaemias. Cornea. 1989;8:75–79. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
