

Sequence and structure continuity of evolutionary importance improves protein functional site discovery and annotation
- PMID: 20506260
- PMCID: PMC2974822
- DOI: 10.1002/pro.406
Sequence and structure continuity of evolutionary importance improves protein functional site discovery and annotation
Abstract
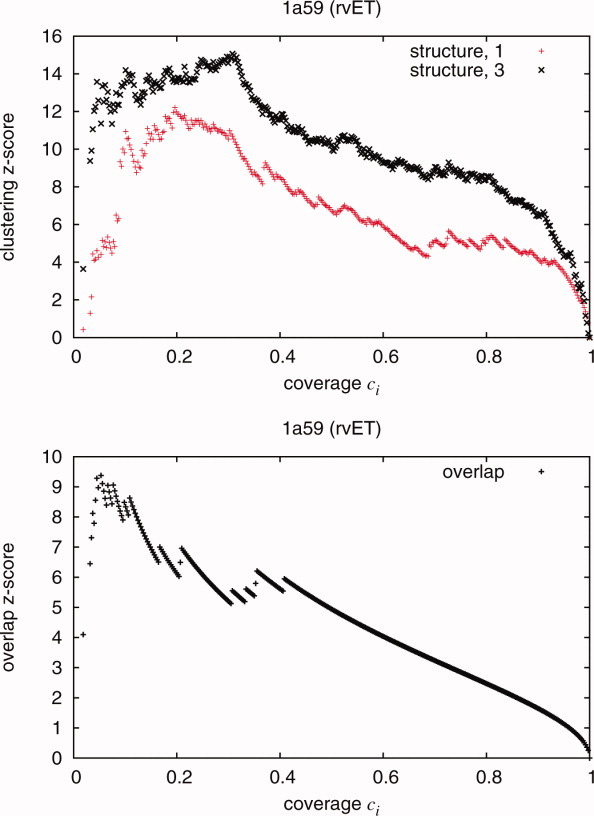
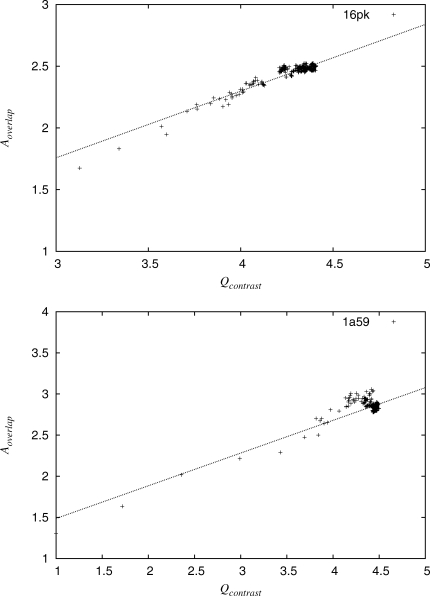
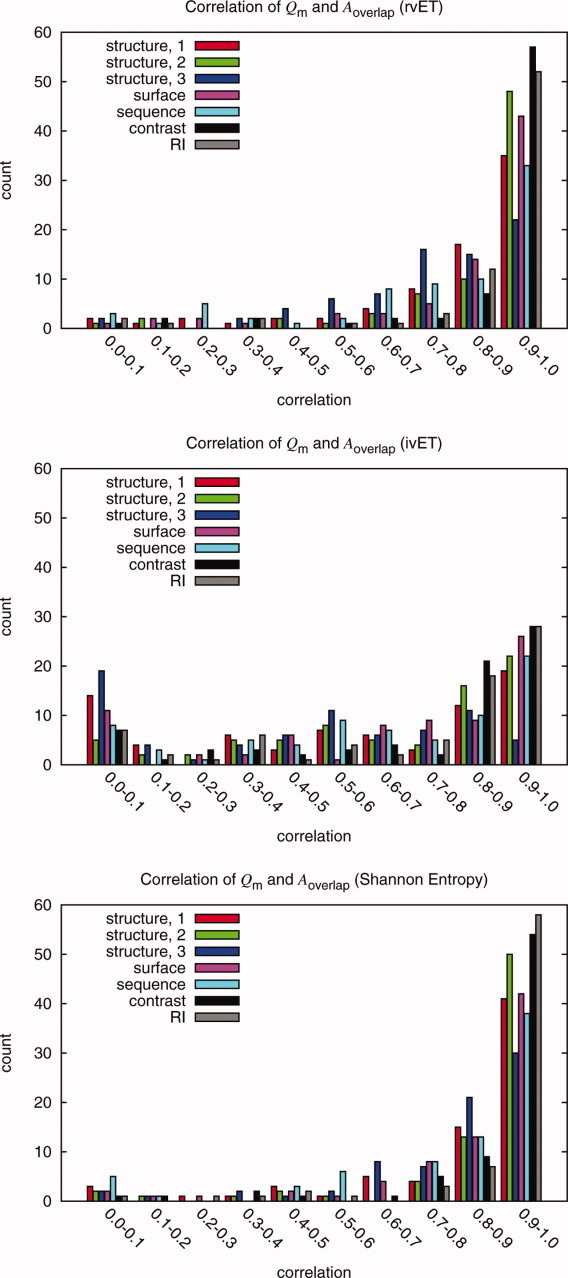
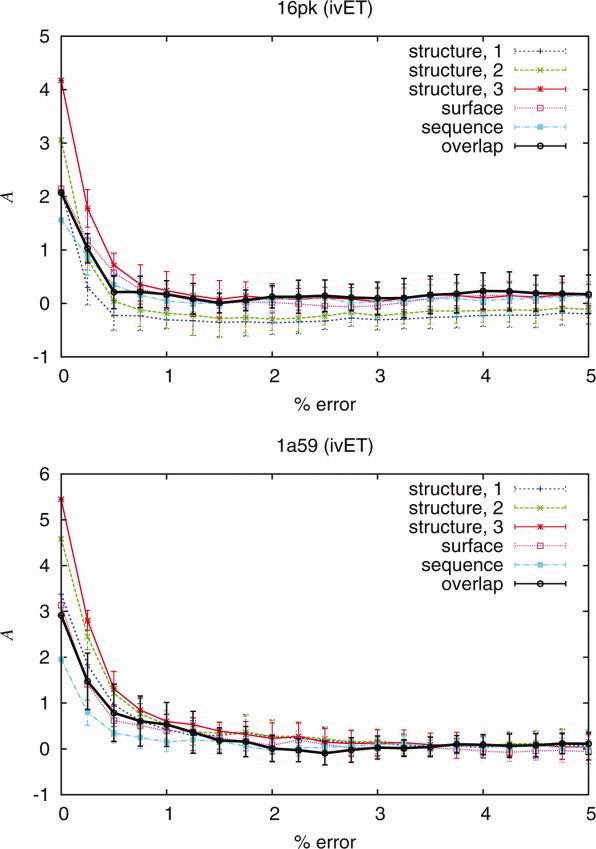
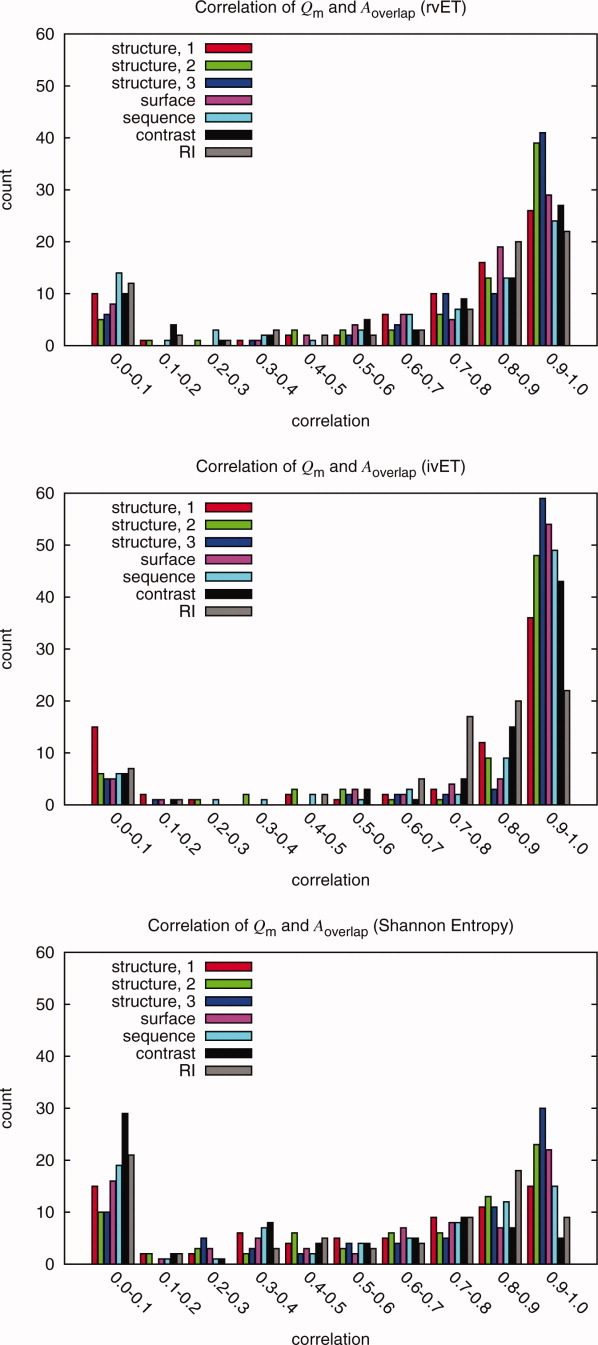
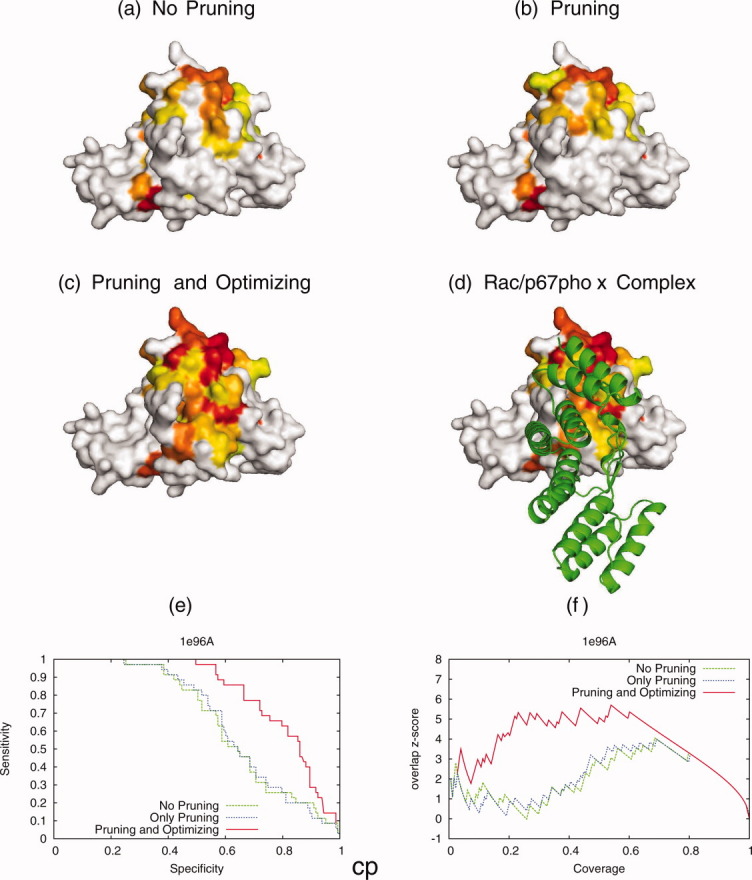
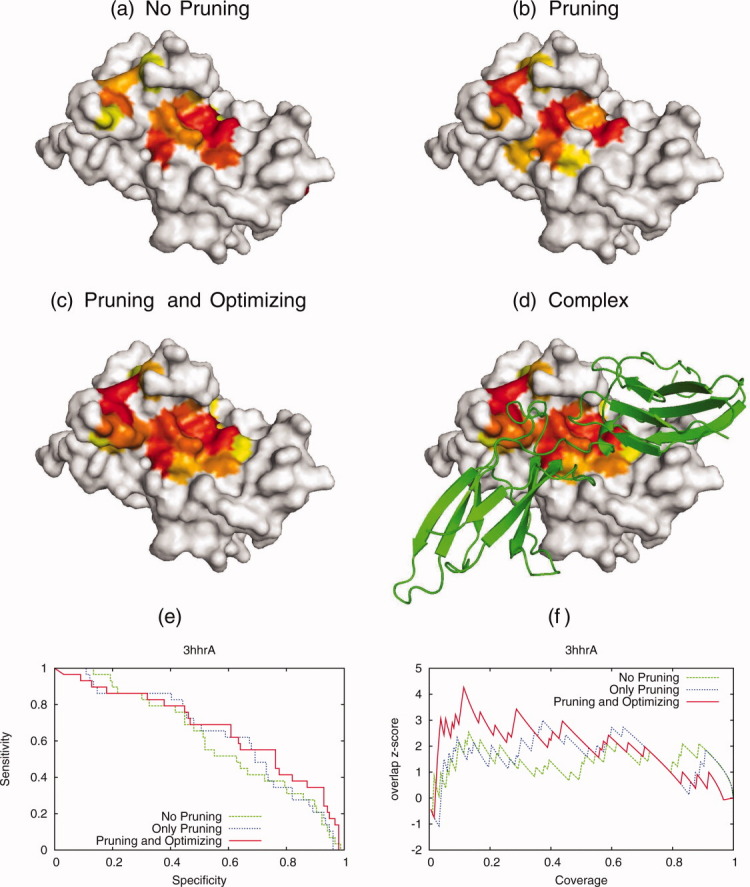
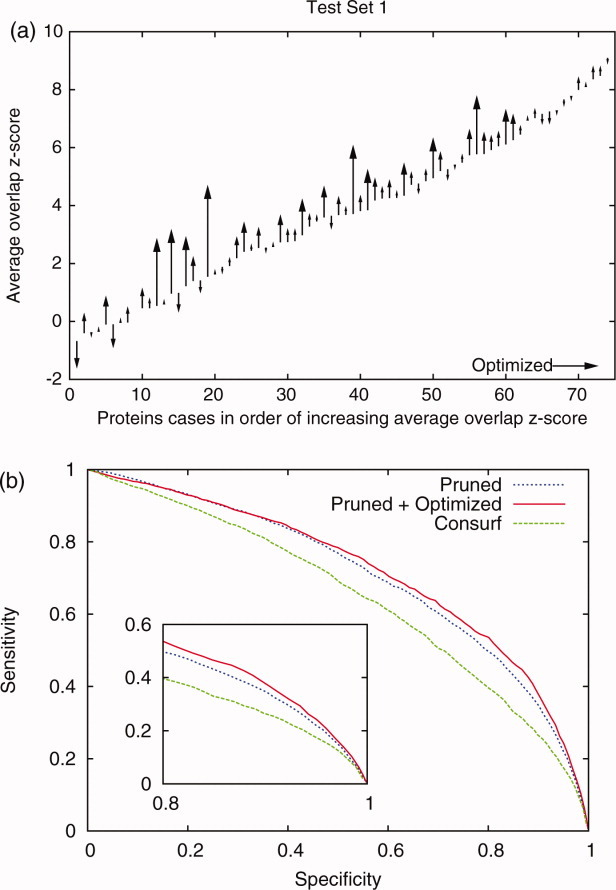
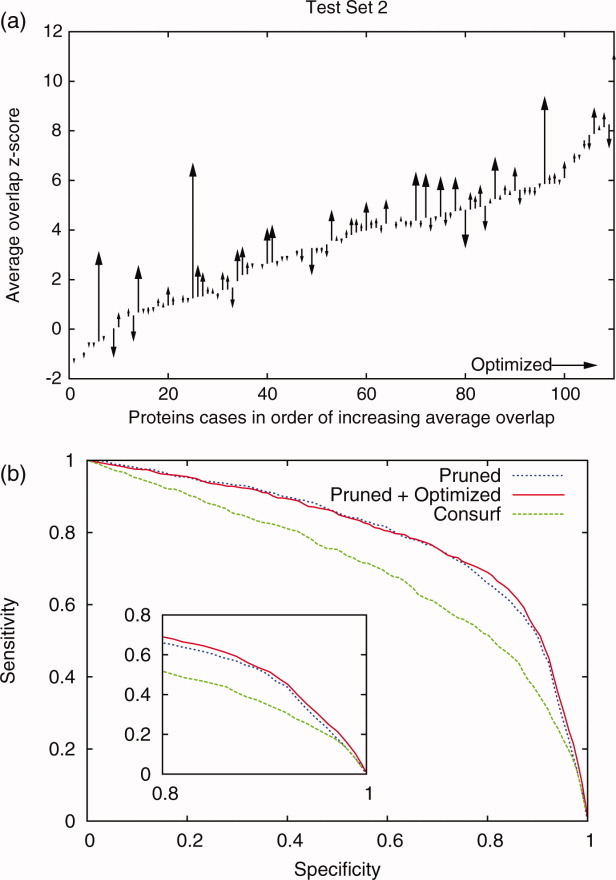
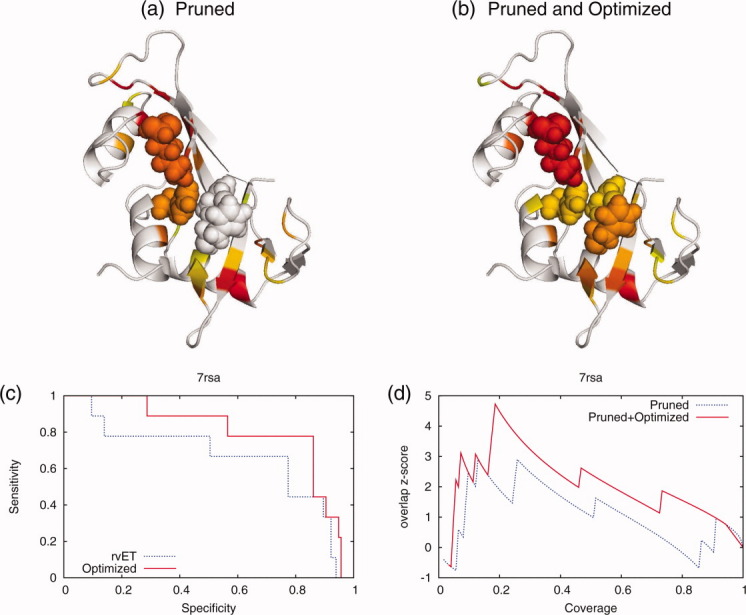
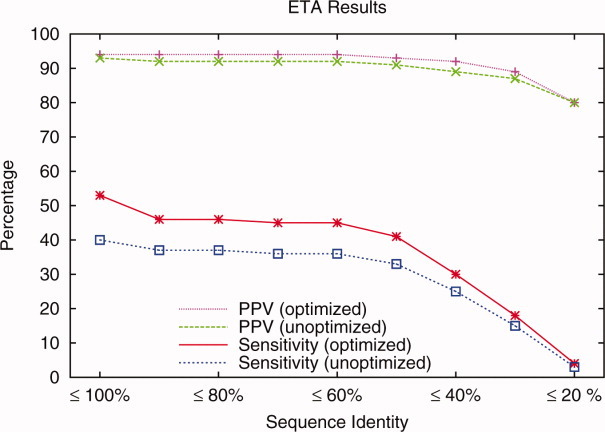
Protein functional sites control most biological processes and are important targets for drug design and protein engineering. To characterize them, the evolutionary trace (ET) ranks the relative importance of residues according to their evolutionary variations. Generally, top-ranked residues cluster spatially to define evolutionary hotspots that predict functional sites in structures. Here, various functions that measure the physical continuity of ET ranks among neighboring residues in the structure, or in the sequence, are shown to inform sequence selection and to improve functional site resolution. This is shown first, in 110 proteins, for which the overlap between top-ranked residues and actual functional sites rose by 8% in significance. Then, on a structural proteomic scale, optimized ET led to better 3D structure-function motifs (3D templates) and, in turn, to enzyme function prediction by the Evolutionary Trace Annotation (ETA) method with better sensitivity of (40% to 53%) and positive predictive value (93% to 94%). This suggests that the similarity of evolutionary importance among neighboring residues in the sequence and in the structure is a universal feature of protein evolution. In practice, this yields a tool for optimizing sequence selections for comparative analysis and, via ET, for better predictions of functional site and function. This should prove useful for the efficient mutational redesign of protein function and for pharmaceutical targeting.
Figures
References
-
- Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8:995–1005. - PubMed
-
- Laskowski RA, Thornton JM. Understanding the molecular machinery of genetics through 3D structures. Nat Rev Genet. 2008;9:141–145. - PubMed
-
- Hardy JA, Wells J. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:706–715. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
