

Chromodomain proteins in development: lessons from CHARGE syndrome
- PMID: 20507341
- PMCID: PMC3097394
- DOI: 10.1111/j.1399-0004.2010.01446.x
Chromodomain proteins in development: lessons from CHARGE syndrome
Abstract
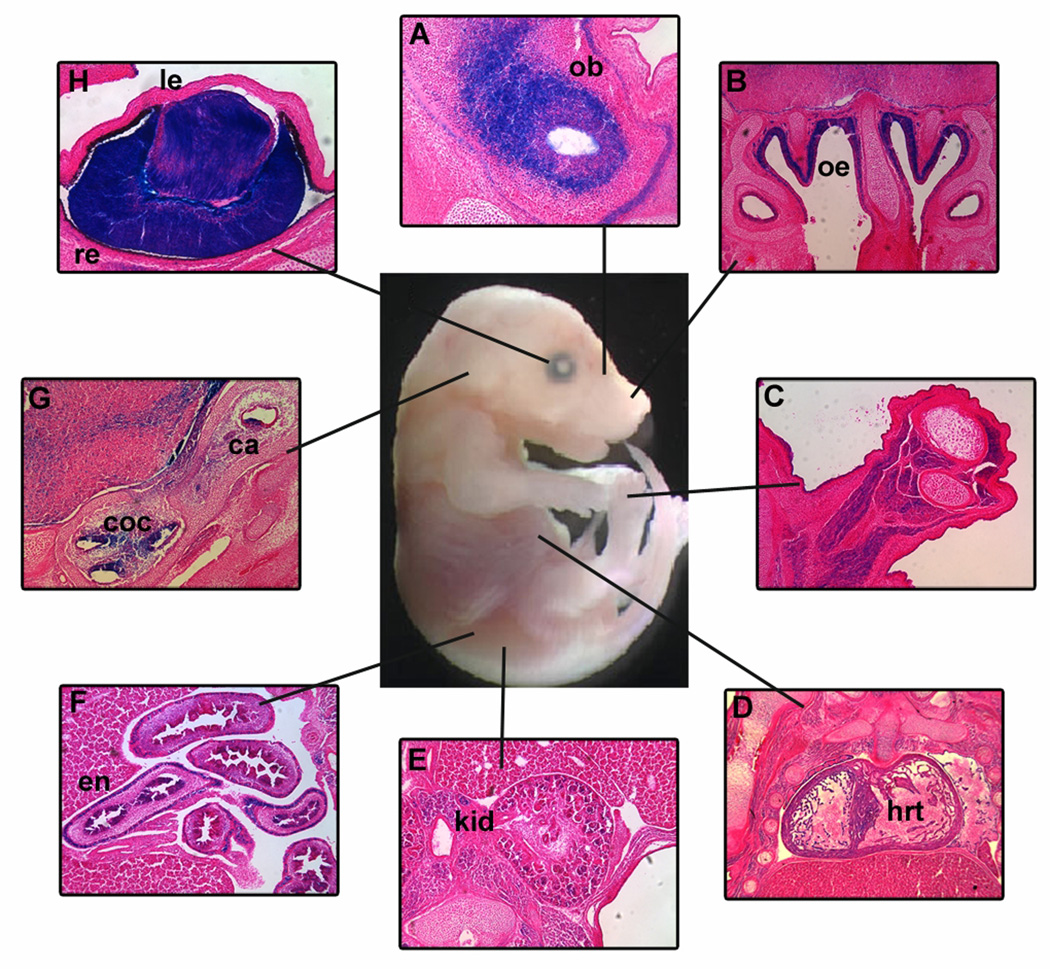
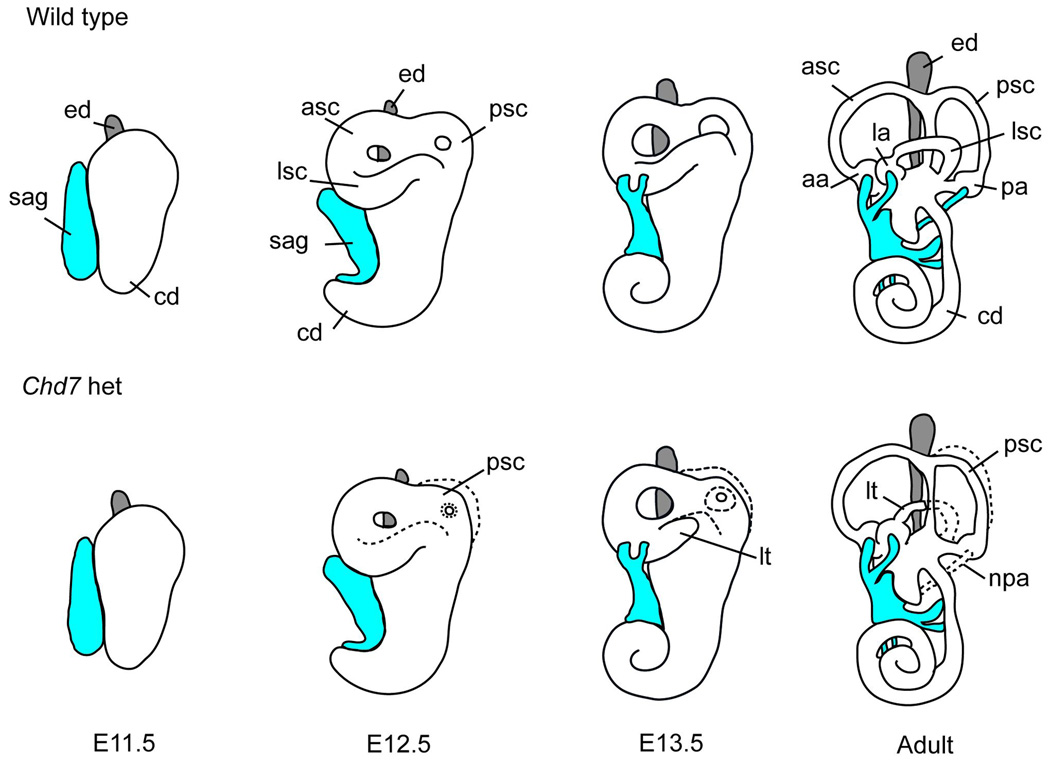
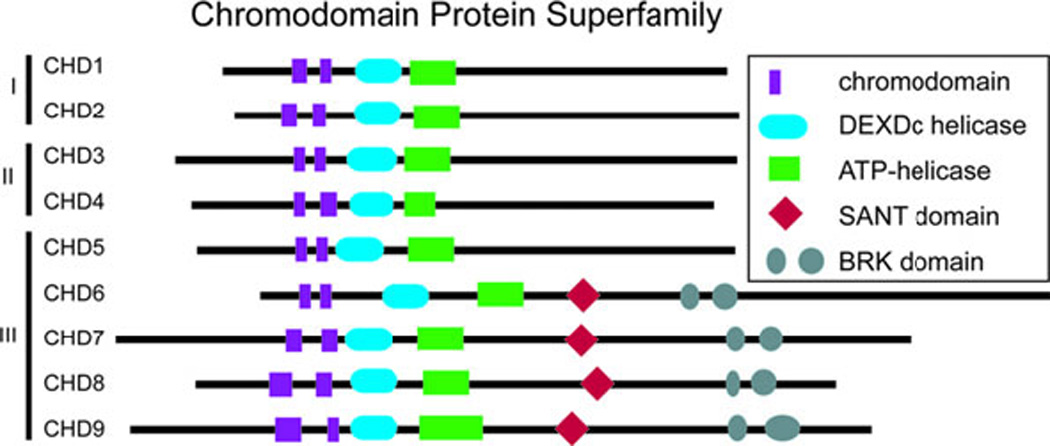
In humans, heterozygous mutations in the adenosine triphosphate-dependent chromatin remodeling gene CHD7 cause CHARGE syndrome, a common cause of deaf-blindness, balance disorders, congenital heart malformations, and olfactory dysfunction with an estimated incidence of approximately 1 in 10,000 newborns. The clinical features of CHARGE in humans and mice are highly variable and incompletely penetrant, and most mutations appear to result in haploinsufficiency of functional CHD7 protein. Mice with heterozygous loss of function mutations in Chd7 are a good model for CHARGE syndrome, and analyses of mouse mutant phenotypes have begun to clarify a role for CHD7 during development and into adulthood. Chd7 heterozygous mutant mice have postnatal delayed growth, inner ear malformations, anosmia/hyposmia, and craniofacial defects, and Chd7 homozygous mutants are embryonic lethal. A central question in developmental biology is how chromodomain proteins like CHD7 regulate important developmental processes, and whether they directly activate or repress downstream gene transcription or act more globally to alter chromatin structure and/or function. CHD7 is expressed in a wide variety of tissues during development, suggesting that it has tissue-specific and developmental stage-specific roles. Here, we review recent and ongoing analyses of CHD7 function in mouse models and cell-based systems. These studies explore tissue-specific effects of CHD7 deficiency, known CHD7 interacting proteins, and downstream target sites for CHD7 binding. CHD7 is emerging as a critical regulator of important developmental processes in organs affected by human CHARGE syndrome.
Conflict of interest statement
Figures


References
-
- Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr. 1979;95:395–398. - PubMed
-
- Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, Oki H, Nanao K, Moriyama N, Oku S, Hasegawa T, Takahashi T, Fukushima Y, Kawame H, Kosaki K. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148:410–414. - PubMed
-
- Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BB, Brunner HG, Hoefsloot LH, van Ravenswaaij CM. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43:306–314. - PMC - PubMed
-
- Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SL, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW. Spectrum of CHD7 Mutations in 110 Individuals with CHARGE Syndrome and Genotype-Phenotype Correlation. Am J Hum Genet. 2006;78:303–314. - PMC - PubMed
-
- Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Bitach T. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43:211–217. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
