

Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase
- PMID: 20507988
- PMCID: PMC2915725
- DOI: 10.1074/jbc.M110.147843
Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase
Abstract
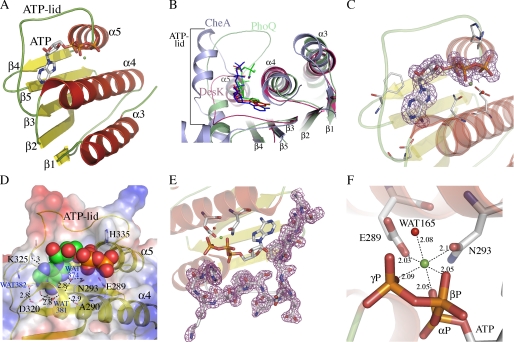
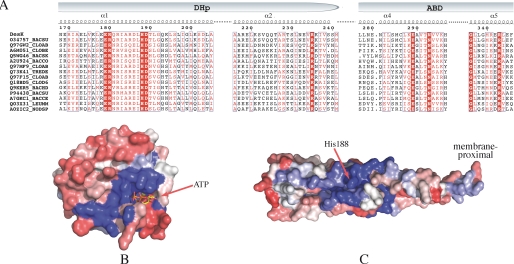
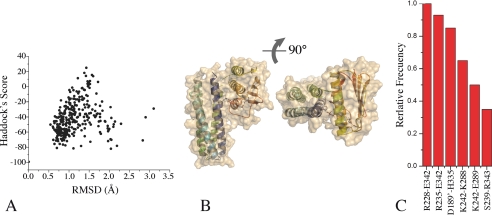
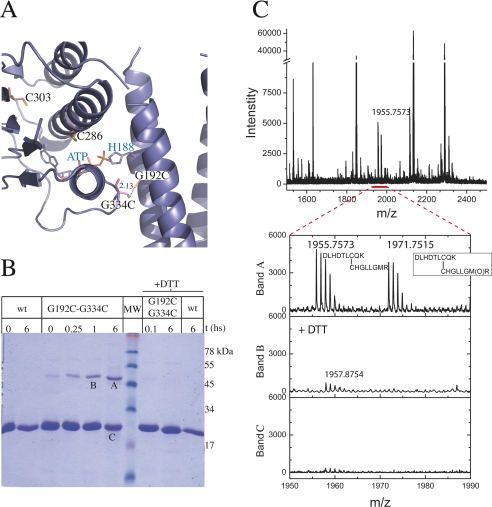
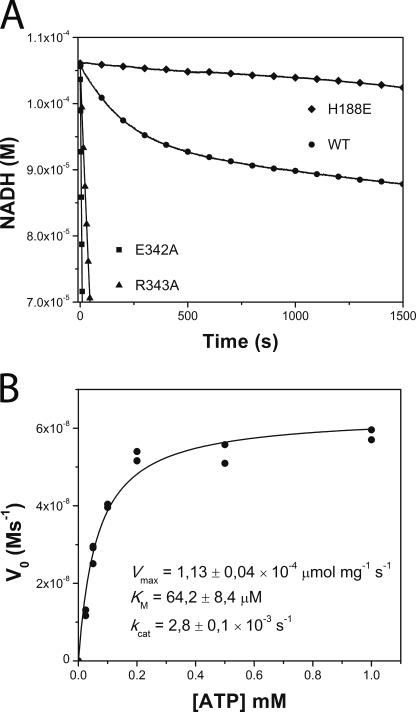
DesK is a sensor histidine kinase (HK) that allows Bacillus subtilis to respond to cold shock, triggering the adaptation of membrane fluidity via transcriptional control of a fatty acid desaturase. It belongs to the HK family HPK7, which includes the nitrogen metabolism regulators NarX/Q and the antibiotic sensor LiaS among other important sensor kinases. Structural information on different HK families is still scarce and several questions remain, particularly concerning the molecular features that determine HK specificity during its catalytic autophosphorylation and subsequent response-regulator phosphotransfer reactions. To analyze the ATP-binding features of HPK7 HKs and dissect their mechanism of autophosphorylation at the molecular level, we have studied DesK in complex with ATP using high resolution structural approaches in combination with biochemical studies. We report the first crystal structure of an HK in complex with its natural nucleotidic substrate. The general fold of the ATP-binding domain of DesK is conserved, compared with well studied members of other families. Yet, DesK displays a far more compact structure at the ATP-binding pocket: the ATP lid loop is much shorter with no secondary structural organization and becomes ordered upon ATP loading. Sequence conservation mapping onto the molecular surface, semi-flexible protein-protein docking simulations, and structure-based point mutagenesis allow us to propose a specific domain-domain geometry during autophosphorylation catalysis. Supporting our hypotheses, we have been able to trap an autophosphorylating intermediate state, by protein engineering at the predicted domain-domain interaction surface.
Figures
References
-
- Wolanin P. M., Thomason P. A., Stock J. B. (2002) Genome Biol. 3, REVIEWS3013
-
- Laub M. T., Goulian M. (2007) Annu. Rev. Genet 41, 121–145 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
