

A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants
- PMID: 20512113
- PMCID: PMC2905243
- DOI: 10.1038/emboj.2010.104
A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants
Abstract
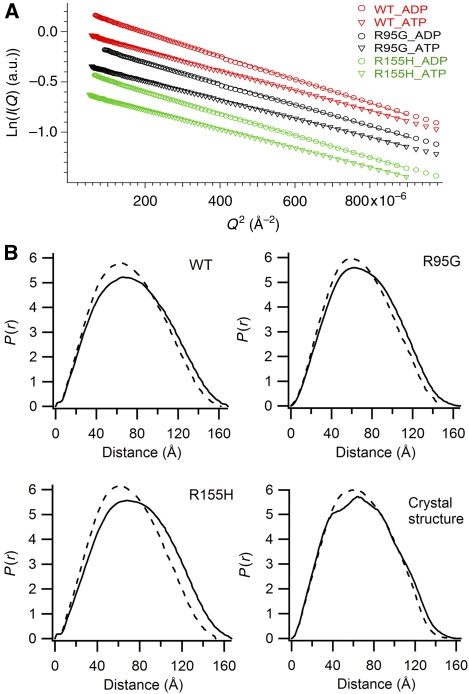
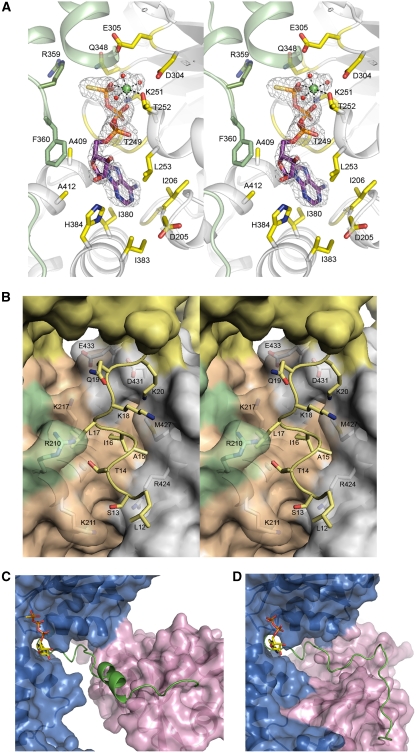
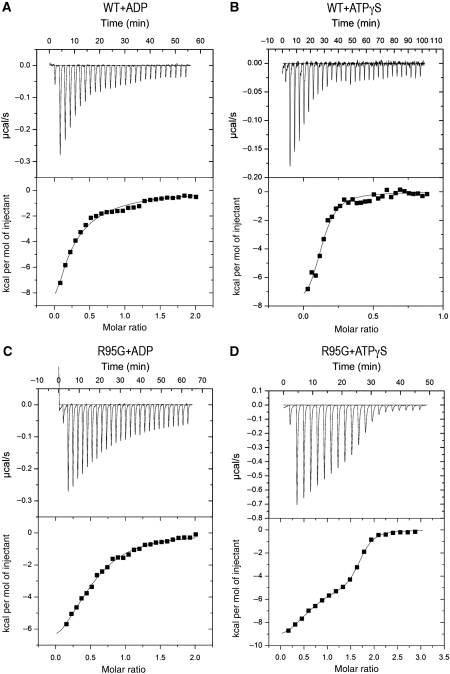
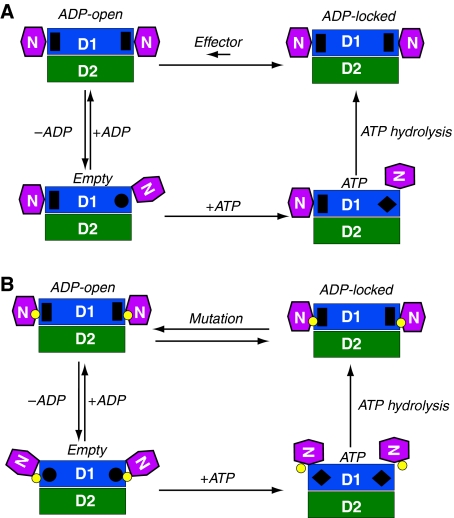
Mutations in p97, a major cytosolic AAA (ATPases associated with a variety of cellular activities) chaperone, cause inclusion body myopathy associated with Paget's disease of the bone and frontotemporal dementia (IBMPFD). IBMPFD mutants have single amino-acid substitutions at the interface between the N-terminal domain (N-domain) and the adjacent AAA domain (D1), resulting in a reduced affinity for ADP. The structures of p97 N-D1 fragments bearing IBMPFD mutations adopt an atypical N-domain conformation in the presence of Mg(2+).ATPgammaS, which is reversible by ADP, showing for the first time the nucleotide-dependent conformational change of the N-domain. The transition from the ADP- to the ATPgammaS-bound state is accompanied by a loop-to-helix conversion in the N-D1 linker and by an apparent re-ordering in the N-terminal region of p97. X-ray scattering experiments suggest that wild-type p97 subunits undergo a similar nucleotide-dependent N-domain conformational change. We propose that IBMPFD mutations alter the timing of the transition between nucleotide states by destabilizing the ADP-bound form and consequently interfere with the interactions between the N-domains and their substrates.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Beuron F, Flynn TC, Ma J, Kondo H, Zhang X, Freemont PS (2003) Motions and negative cooperativity between p97 domains revealed by cryo-electron microscopy and quantised elastic deformational model. J Mol Biol 327: 619–629 - PubMed
-
- Dai RM, Li CC (2001) Valosin-containg protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 3: 740–744 - PubMed
-
- Davies JM, Tsuruta H, May AP, Weis WI (2005) Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-ray scattering. Structure 13: 183–195 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
