

Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes
- PMID: 20512146
- PMCID: PMC2894012
- DOI: 10.1038/ng.594
Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes
Abstract
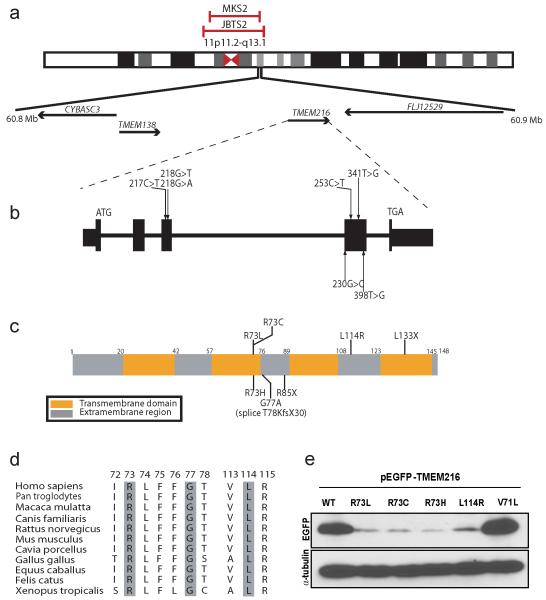
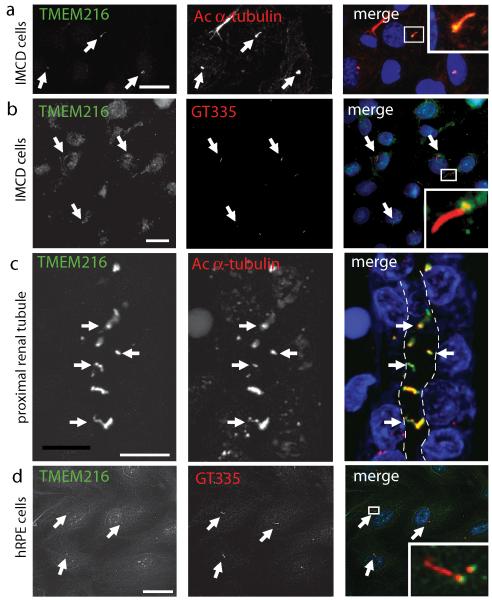
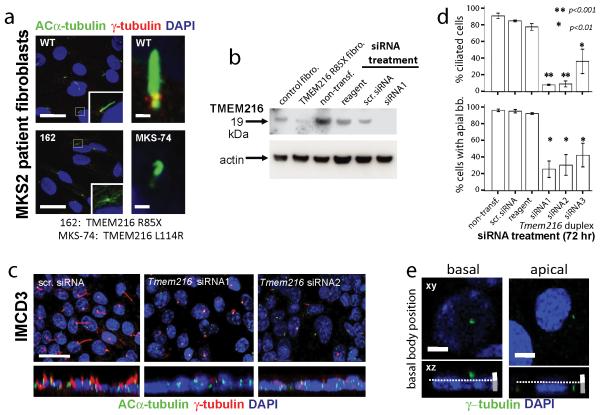
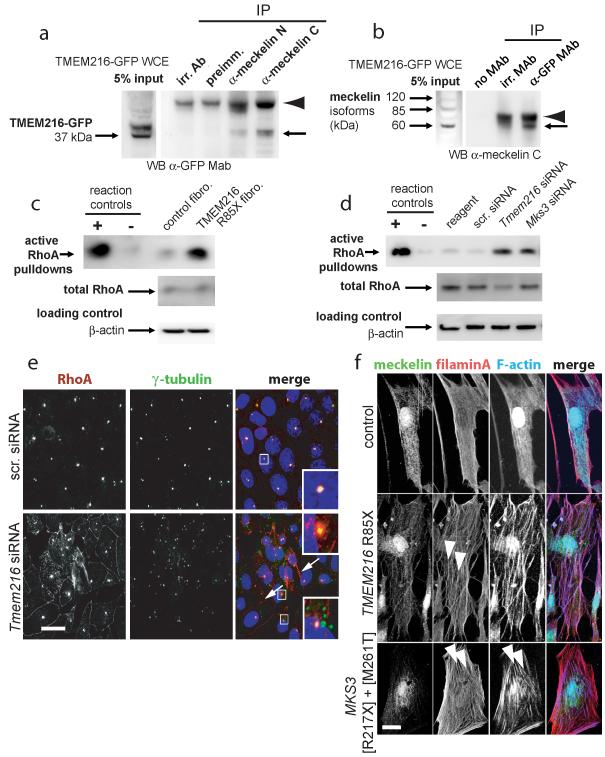
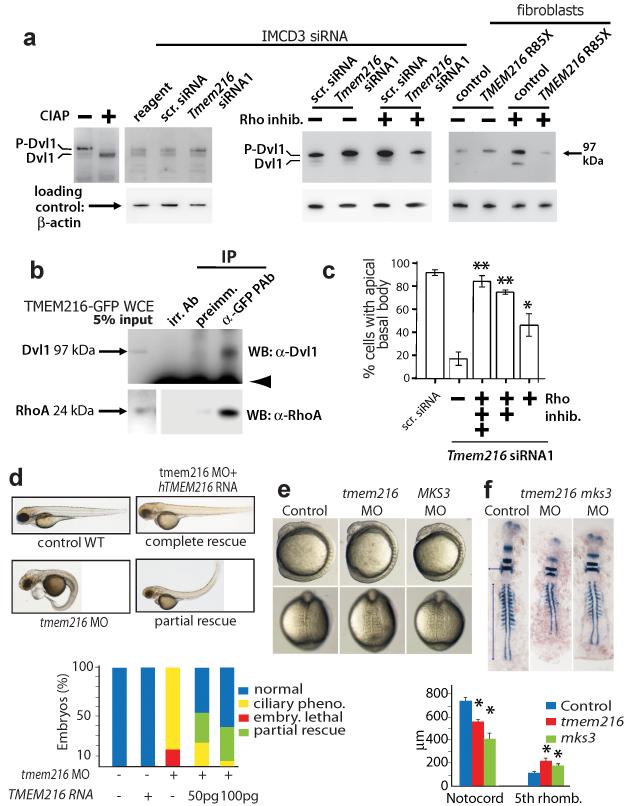
Joubert syndrome (JBTS), related disorders (JSRDs) and Meckel syndrome (MKS) are ciliopathies. We now report that MKS2 and CORS2 (JBTS2) loci are allelic and caused by mutations in TMEM216, which encodes an uncharacterized tetraspan transmembrane protein. Individuals with CORS2 frequently had nephronophthisis and polydactyly, and two affected individuals conformed to the oro-facio-digital type VI phenotype, whereas skeletal dysplasia was common in fetuses affected by MKS. A single G218T mutation (R73L in the protein) was identified in all cases of Ashkenazi Jewish descent (n=10). TMEM216 localized to the base of primary cilia, and loss of TMEM216 in mutant fibroblasts or after knockdown caused defective ciliogenesis and centrosomal docking, with concomitant hyperactivation of RhoA and Dishevelled. TMEM216 formed a complex with Meckelin, which is encoded by a gene also mutated in JSRDs and MKS. Disruption of tmem216 expression in zebrafish caused gastrulation defects similar to those in other ciliary morphants. These data implicate a new family of proteins in the ciliopathies and further support allelism between ciliopathy disorders.
Figures
Comment in
-
TMEM216 joins its ciliary cousins in ciliopathies.Clin Genet. 2011 Jan;79(1):45-7. doi: 10.1111/j.1399-0004.2010.01556_2.x. Epub 2010 Oct 12. Clin Genet. 2011. PMID: 21029074 No abstract available.
References
-
- Valente EM, et al. Distinguishing the four genetic causes of Joubert syndrome-related disorders. Ann Neurol. 2005;57:513–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G0700073/MRC_/Medical Research Council/United Kingdom
- HHMI/Howard Hughes Medical Institute/United States
- R01 NS048453/NS/NINDS NIH HHS/United States
- R01 DK068306/DK/NIDDK NIH HHS/United States
- GGP08145/TI_/Telethon/Italy
- R01 NS052455/NS/NINDS NIH HHS/United States
- R01 DK075972/DK/NIDDK NIH HHS/United States
- F32 DK079541/DK/NIDDK NIH HHS/United States
- R01 HD042601/HD/NICHD NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- P30NS047101/NS/NINDS NIH HHS/United States
- G0700073(81627)/MRC_/Medical Research Council/United Kingdom
- R01 HD04260/HD/NICHD NIH HHS/United States
- R01 NS04843/NS/NINDS NIH HHS/United States
- P30 NS047101/NS/NINDS NIH HHS/United States
- R01 DK072301/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
