

New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish
- PMID: 20512159
- PMCID: PMC2987448
- DOI: 10.1038/ejhg.2010.79
New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish
Abstract
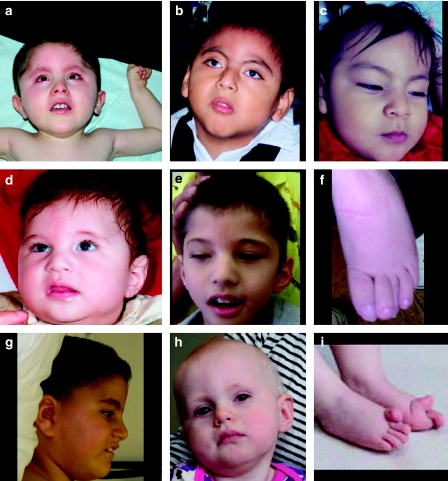
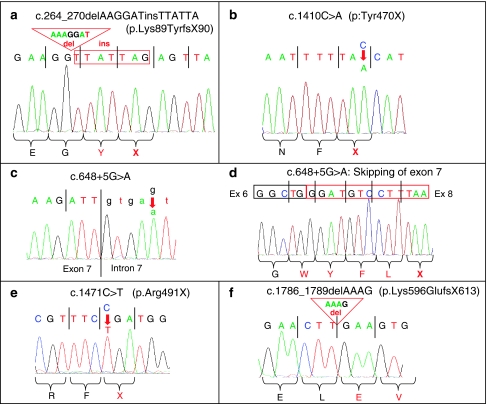
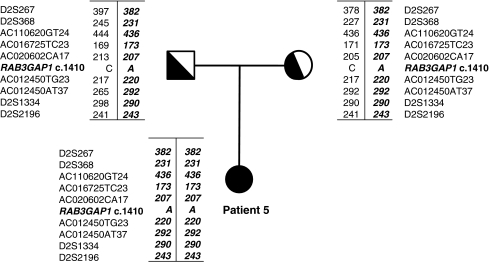
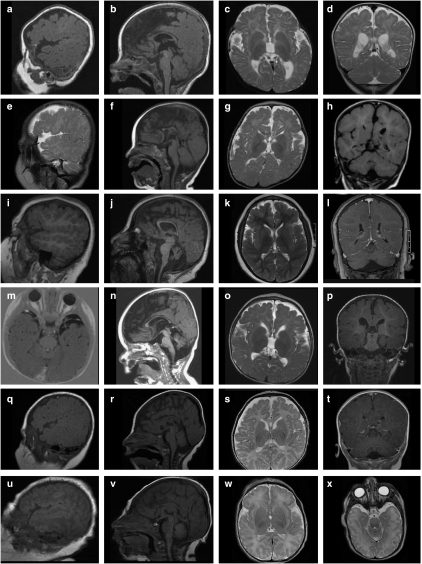
Warburg Micro Syndrome is a rare, autosomal recessive syndrome characterized by microcephaly, microphthalmia, microcornia, congenital cataracts, optic atrophy, cortical dysplasia, in particular corpus callosum hypoplasia, severe mental retardation, spastic diplegia, and hypogonadism. We have found five new mutations in the RAB3GAP1 gene in seven patients with suspected Micro Syndrome from families with Turkish, Palestinian, Danish, and Guatemalan backgrounds. A thorough clinical investigation of the patients has allowed the delineation of symptoms that are consistently present in the patients and may aid the differential diagnosis of Micro Syndrome for patients in the future. All patients had postnatal microcephaly, micropthalmia, microcornia, bilateral congenital cataracts, short palpebral fissures, optic atrophy, severe mental retardation, and congenital hypotonia with subsequent spasticity. Only one patient had microcephaly at birth, highlighting the fact that congenital microcephaly is not a consistent feature of Micro syndrome. Analysis of the brain magnetic resonance imagings (MRIs) revealed a consistent pattern of polymicrogyria in the frontal and parietal lobes, wide sylvian fissures, a thin hypoplastic corpus callosum, and increased subdural spaces. All patients were homozygous for the mutations detected and all mutations were predicted to result in a truncated RAB3GAP1 protein. The analysis of nine polymorphic markers flanking the RAB3GAP1 gene showed that the mutation c.1410C>A (p.Tyr470X), for which a Danish patient was homozygous, occurred on a haplotype that is shared by the unrelated heterozygous parents of the patient. This suggests a possible founder effect for this mutation in the Danish population.
Figures
References
-
- Derbent M, Agras PI, Gedik S, Oto S, Alehan F, Saatci U. Congenital cataract, microphthalmia, hypoplasia of corpus callosum and hypogenitalism: report and review of Micro syndrome. Am J Med Genet A. 2004;128A:232–234. - PubMed
-
- Graham JM, Jr, Hennekam R, Dobyns WB, Roeder E, Busch D. MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet A. 2004;128A:235–245. - PubMed
-
- Warburg M, Sjo O, Fledelius HC, Pedersen SA. Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child. 1993;147:1309–1312. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
