

Splicing of the Survival Motor Neuron genes and implications for treatment of SMA
- PMID: 20515750
- PMCID: PMC2921696
- DOI: 10.2741/3670
Splicing of the Survival Motor Neuron genes and implications for treatment of SMA
Abstract
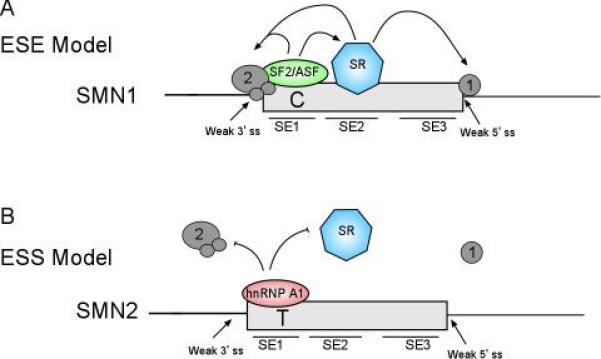
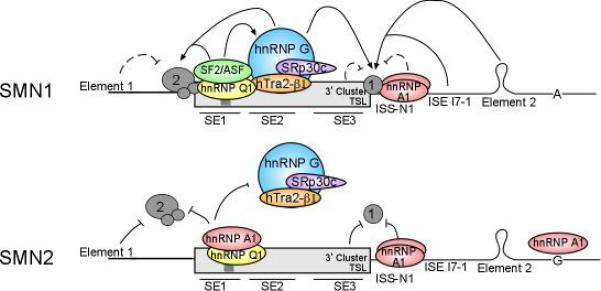
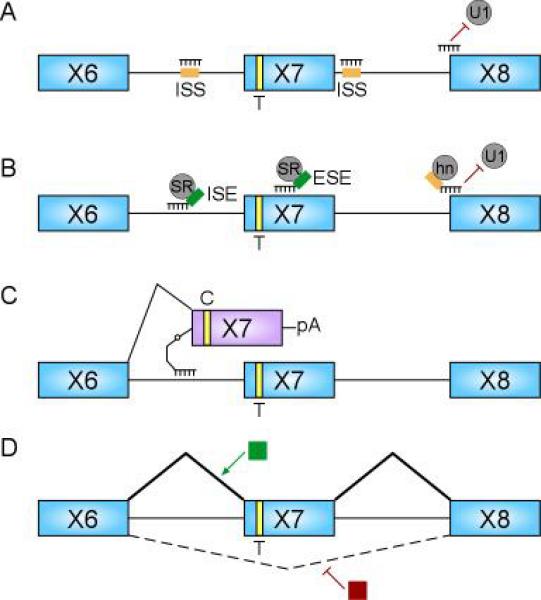
Proximal spinal muscular atrophy (SMA) is a neuromuscular disease caused by low levels of the survival motor neuron (SMN) protein. The reduced SMN levels are due to loss of the survival motor neuron-1 (SMN1) gene. Humans carry a nearly identical SMN2 gene that generates a truncated protein, due to a C to T nucleotide alteration in exon 7 that leads to inefficient RNA splicing of exon 7. This exclusion of SMN exon 7 is central to the onset of the SMA disease, however, this offers a unique therapeutic intervention in which corrective splicing of the SMN2 gene would restore SMN function. Exon 7 splicing is regulated by a number of exonic and intronic splicing regulatory sequences and trans-factors that bind them. A better understanding of the way SMN pre-mRNA is spliced has lead to the development of targeted therapies aimed at correcting SMN2 splicing. As therapeutics targeted toward correction of SMN2 splicing continue to be developed available SMA mouse models can be utilized in validating their potential in disease treatment.
Figures
Similar articles
-
5-(N-ethyl-N-isopropyl)-amiloride enhances SMN2 exon 7 inclusion and protein expression in spinal muscular atrophy cells.Ann Neurol. 2008 Jan;63(1):26-34. doi: 10.1002/ana.21241. Ann Neurol. 2008. PMID: 17924536
-
Heat increases full-length SMN splicing: promise for splice-augmenting therapies for SMA.Hum Genet. 2022 Feb;141(2):239-256. doi: 10.1007/s00439-021-02408-7. Epub 2022 Jan 28. Hum Genet. 2022. PMID: 35088120 Free PMC article.
-
Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes.Adv Neurobiol. 2018;20:31-61. doi: 10.1007/978-3-319-89689-2_2. Adv Neurobiol. 2018. PMID: 29916015 Free PMC article. Review.
-
Alternative splicing in spinal muscular atrophy underscores the role of an intron definition model.RNA Biol. 2011 Jul-Aug;8(4):600-6. doi: 10.4161/rna.8.4.16224. Epub 2011 Jul 1. RNA Biol. 2011. PMID: 21654213 Free PMC article.
-
Therapeutics that directly increase SMN expression to treat spinal muscular atrophy.Drug News Perspect. 2010 Oct;23(8):475-82. doi: 10.1358/dnp.2010.23.8.1507295. Drug News Perspect. 2010. PMID: 21031163 Review.
Cited by
-
Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production.Glia. 2013 Sep;61(9):1418-1428. doi: 10.1002/glia.22522. Epub 2013 Jul 10. Glia. 2013. PMID: 23839956 Free PMC article.
-
Transgenic and knockout analyses of Masculinizer and doublesex illuminated the unique functions of doublesex in germ cell sexual development of the silkworm, Bombyx mori.BMC Dev Biol. 2020 Sep 21;20(1):19. doi: 10.1186/s12861-020-00224-2. BMC Dev Biol. 2020. PMID: 32957956 Free PMC article.
-
Characterization of the RNA recognition mode of hnRNP G extends its role in SMN2 splicing regulation.Nucleic Acids Res. 2014 Jun;42(10):6659-72. doi: 10.1093/nar/gku244. Epub 2014 Apr 1. Nucleic Acids Res. 2014. PMID: 24692659 Free PMC article.
-
Hypoxia is a modifier of SMN2 splicing and disease severity in a severe SMA mouse model.Hum Mol Genet. 2012 Oct 1;21(19):4301-13. doi: 10.1093/hmg/dds263. Epub 2012 Jul 3. Hum Mol Genet. 2012. PMID: 22763238 Free PMC article.
-
SMN-inducing compounds for the treatment of spinal muscular atrophy.Future Med Chem. 2012 Oct;4(16):2067-84. doi: 10.4155/fmc.12.131. Future Med Chem. 2012. PMID: 23157239 Free PMC article. Review.
References
-
- Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6(8):1205–14. - PubMed
-
- Le TT, Coovert DD, Monani UR, Morris GE, Burghes AH. The survival motor neuron (SMN) protein: effect of exon loss and mutation on protein localization. Neurogenetics. 2000;3(1):7–16. - PubMed
-
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14(6):845–57. - PubMed
-
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–9. - PubMed
-
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9(3):333–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
