

The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition
- PMID: 20516069
- PMCID: PMC2930675
- DOI: 10.1074/jbc.M109.092304
The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition
Abstract
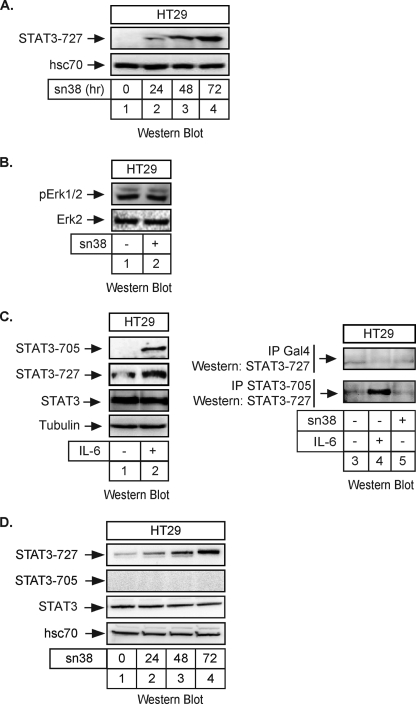
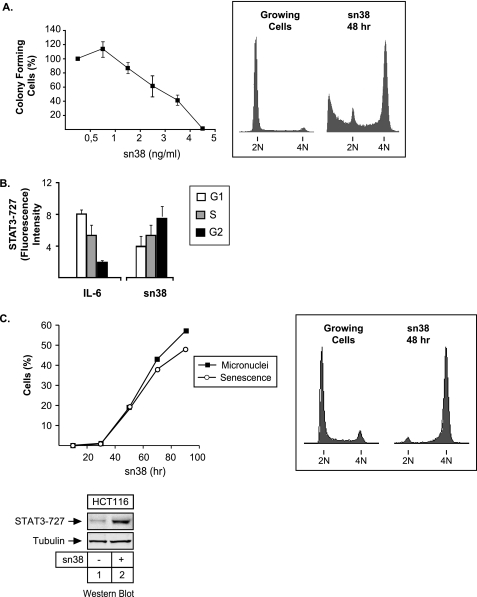
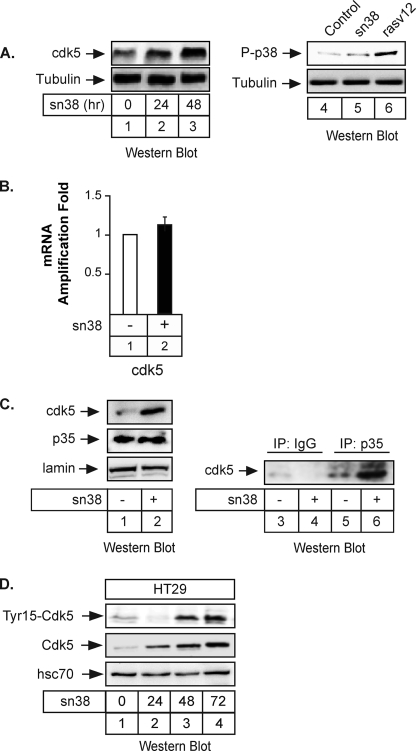
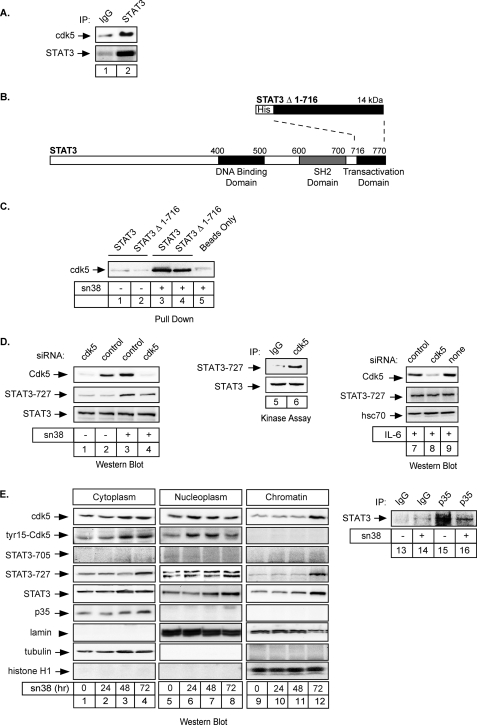
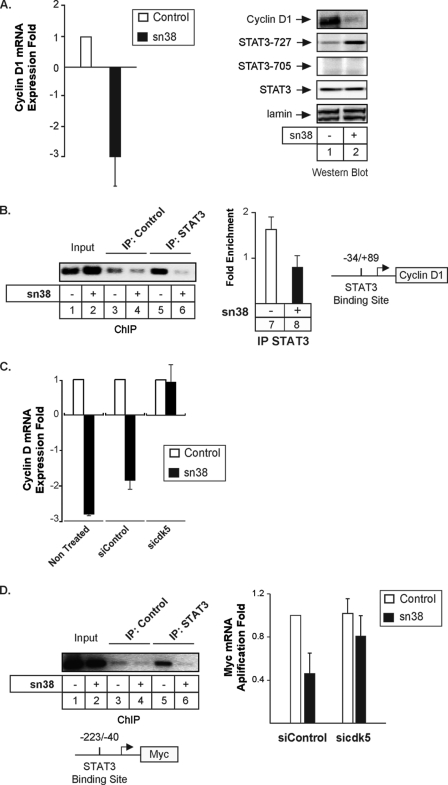
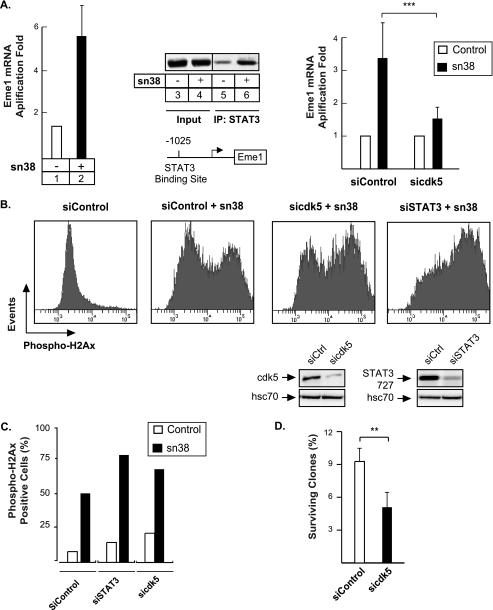
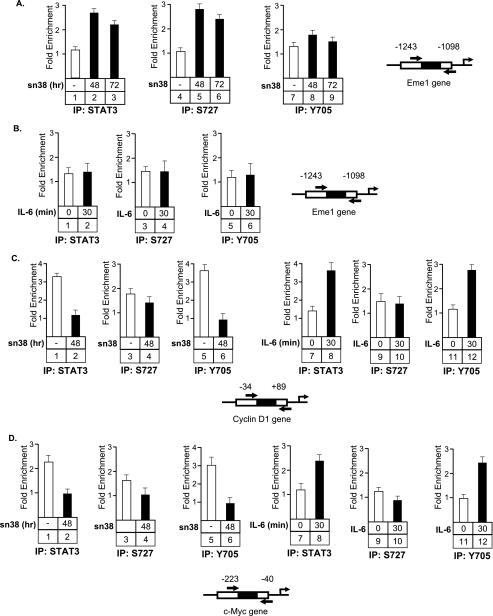
The STAT3 transcription factors are cytoplasmic proteins that induce gene activation in response to growth factor stimulation. Following tyrosine phosphorylation, STAT3 proteins dimerize, translocate to the nucleus, and activate specific target genes involved in cell-cycle progression. Despite its importance in cancer cells, the molecular mechanisms by which this protein is regulated in response to DNA damage remain to be characterized. In this study, we show that STAT3 is activated in response to topoisomerase I inhibition. Following treatment, STAT3 is phosphorylated on its C-terminal serine 727 residue but not on its tyrosine 705 site. We also show that topoisomerase I inhibition induced the up-regulation of the cdk5 kinase, a protein initially described in neuronal stress responses. In co-immunoprecipitations, cdk5 was found to associate with STAT3, and pulldown experiments indicated that it associates with the C-terminal activation domain of STAT3 upon DNA damage. Importantly, the cdk5-STAT3 pathway reduced DNA damage in response to topoisomerase I inhibition through the up-regulation of Eme1, an endonuclease involved in DNA repair. ChIP experiments indicated that STAT3 can be found associated with the Eme1 promoter when phosphorylated only on its serine 727 residue and not on tyrosine 705. We therefore propose that the cdk5-STAT3 oncogenic pathway plays an important role in the expression of DNA repair genes and that these proteins could be used as predictive markers of tumors that will fail to respond to chemotherapy.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
