

p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G(2) DNA damage checkpoint in human cancer cells
- PMID: 20516219
- PMCID: PMC2916406
- DOI: 10.1128/MCB.00949-09
p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G(2) DNA damage checkpoint in human cancer cells
Abstract
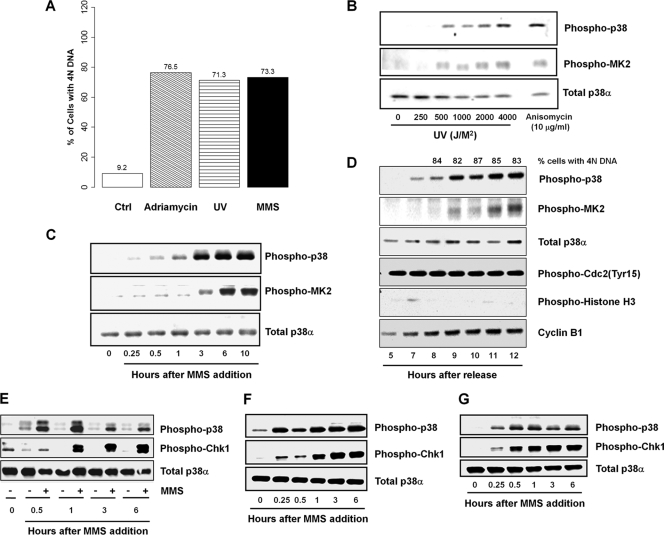
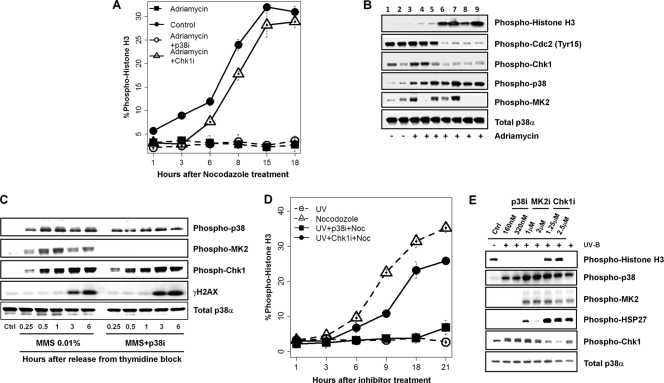
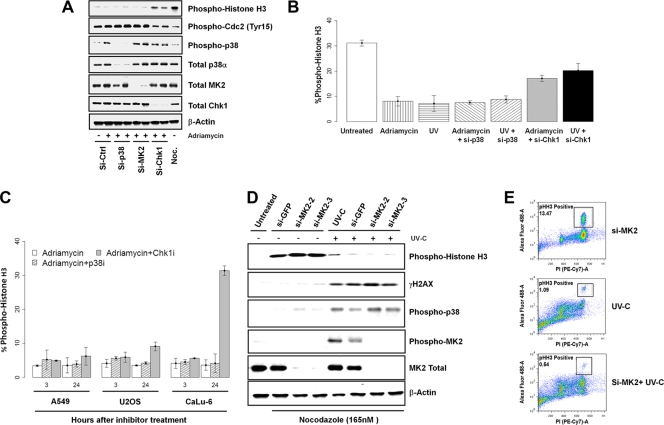
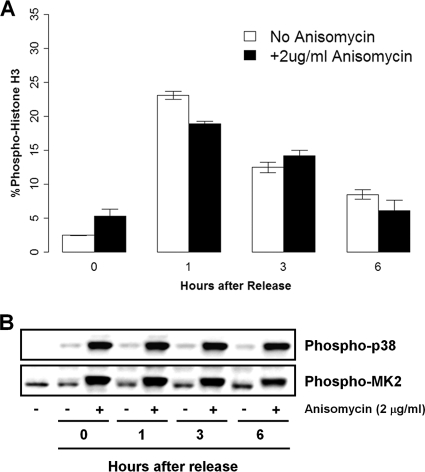
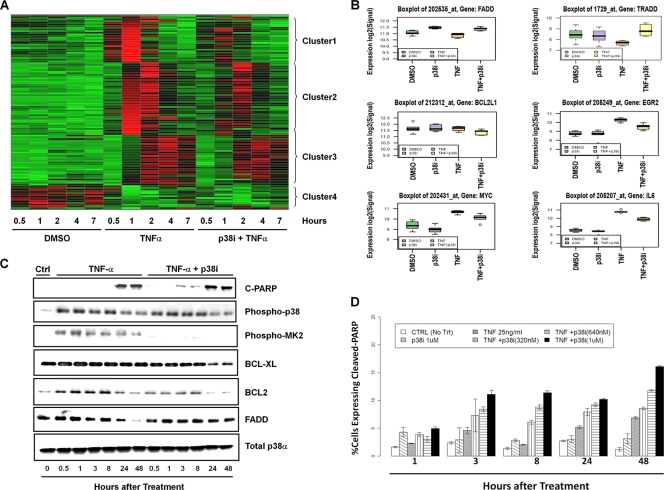
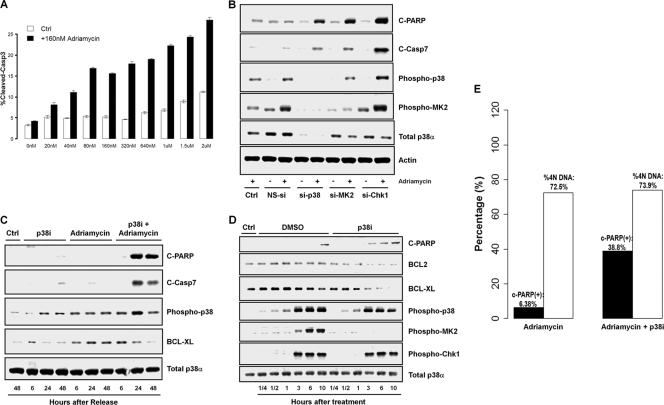
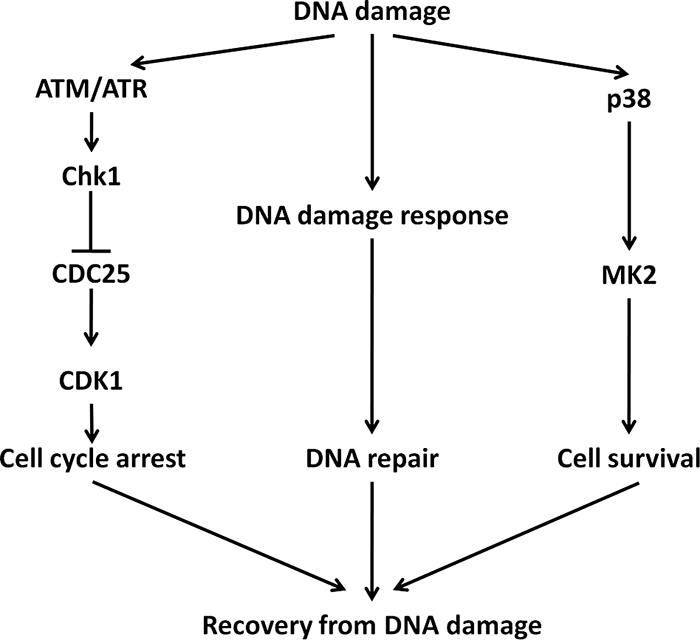
p38 mitogen-activated protein kinase (MAPK) is rapidly activated by stresses and is believed to play an important role in the stress response. While Chk1 is known to mediate G(2) DNA damage checkpoint control, p38 was also reported to have an essential function in this checkpoint control. Here, we have investigated further the roles of p38 and Chk1 in the G(2) DNA damage checkpoint in cancer cells. We find that although p38 activation is strongly induced by DNA damage, its activity is not required for the G(2) DNA damage checkpoint. In contrast, Chk1 kinase is responsible for the execution of G(2) DNA damage checkpoint control in p53-deficient cells. The inhibition of p38 activity has no effect on Chk1 activation and gamma-H2AX expression. Global gene expression profiling of cancer cells in response to tumor necrosis factor alpha (TNF-alpha) revealed that p38 plays a strong prosurvival role through the coordinated downregulation of proapoptotic genes and upregulation of prosurvival genes. We show that the inhibition of p38 activity during G(2) DNA damage checkpoint arrest triggers apoptosis in a p53-independent manner with a concurrent decrease in the level of Bcl2 family proteins. Our results suggest that although p38 MAPK is not required for the G(2) DNA damage checkpoint function, it plays an important prosurvival role during the G(2) DNA damage checkpoint response through the upregulation of the Bcl2 family proteins.
Figures
Similar articles
-
Chk1 inhibition activates p53 through p38 MAPK in tetraploid cancer cells.Cell Cycle. 2008 Jul 1;7(13):1956-61. doi: 10.4161/cc.7.13.6073. Cell Cycle. 2008. PMID: 18642443
-
Arsenic trioxide augments Chk2/p53-mediated apoptosis by inhibiting oncogenic Wip1 phosphatase.J Biol Chem. 2008 Jul 4;283(27):18969-79. doi: 10.1074/jbc.M800560200. Epub 2008 May 15. J Biol Chem. 2008. PMID: 18482988
-
DNA damage dependent activation of checkpoint kinase-1 and mitogen-activated protein kinase-p38 are required in malabaricone C-induced mitochondrial cell death.Biochim Biophys Acta. 2014 Mar;1840(3):1014-27. doi: 10.1016/j.bbagen.2013.11.020. Epub 2013 Nov 27. Biochim Biophys Acta. 2014. PMID: 24291689
-
p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony.Curr Opin Genet Dev. 2002 Feb;12(1):92-7. doi: 10.1016/s0959-437x(01)00270-2. Curr Opin Genet Dev. 2002. PMID: 11790561 Review.
-
The role of p38 MAPK pathway in p53 compromised state and telomere mediated DNA damage response.Mutat Res Genet Toxicol Environ Mutagen. 2018 Dec;836(Pt A):89-97. doi: 10.1016/j.mrgentox.2018.05.018. Epub 2018 May 26. Mutat Res Genet Toxicol Environ Mutagen. 2018. PMID: 30389168 Review.
Cited by
-
The Bidirectional Link Between RNA Cleavage and Polyadenylation and Genome Stability: Recent Insights From a Systematic Screen.Front Genet. 2022 Apr 28;13:854907. doi: 10.3389/fgene.2022.854907. eCollection 2022. Front Genet. 2022. PMID: 35571036 Free PMC article. Review.
-
Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease.Cell Chem Biol. 2017 Aug 17;24(8):1029-1039.e7. doi: 10.1016/j.chembiol.2017.07.011. Cell Chem Biol. 2017. PMID: 28820959 Free PMC article.
-
Cytocidal effects of arenobufagin and hellebrigenin, two active bufadienolide compounds, against human glioblastoma cell line U-87.Int J Oncol. 2018 Dec;53(6):2488-2502. doi: 10.3892/ijo.2018.4567. Epub 2018 Sep 20. Int J Oncol. 2018. PMID: 30272276 Free PMC article.
-
Diversity and versatility of p38 kinase signalling in health and disease.Nat Rev Mol Cell Biol. 2021 May;22(5):346-366. doi: 10.1038/s41580-020-00322-w. Epub 2021 Jan 27. Nat Rev Mol Cell Biol. 2021. PMID: 33504982 Free PMC article. Review.
-
Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways.Biomolecules. 2021 Oct 2;11(10):1444. doi: 10.3390/biom11101444. Biomolecules. 2021. PMID: 34680077 Free PMC article. Review.
References
-
- Blasina, A., J. Hallin, E. Chen, M. E. Arango, E. Kraynov, J. Register, S. Grant, S. Ninkovic, P. Chen, T. Nichols, P. O'Connor, and K. Anderes. 2008. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 7:2394-2404. - PubMed
-
- Bowen, W. P., and P. G. Wylie. 2006. Application of laser-scanning fluorescence microplate cytometry in high content screening. Assay Drug Dev. Technol. 4:209-221. - PubMed
-
- Bulavin, D. V., Y. Higashimoto, I. J. Popoff, W. A. Gaarde, V. Basrur, O. Potapova, E. Appella, and A. J. Fornace, Jr. 2001. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411:102-107. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous