

Clinical progression in Parkinson disease and the neurobiology of axons
- PMID: 20517933
- PMCID: PMC2918373
- DOI: 10.1002/ana.21995
Clinical progression in Parkinson disease and the neurobiology of axons
Abstract
Despite tremendous growth in recent years in our knowledge of the molecular basis of Parkinson disease (PD) and the molecular pathways of cell injury and death, we remain without therapies that forestall disease progression. Although there are many possible explanations for this lack of success, one is that experimental therapeutics to date have not adequately focused on an important component of the disease process, that of axon degeneration. It remains unknown what neuronal compartment, either the soma or the axon, is involved at disease onset, although some have proposed that it is the axons and their terminals that take the initial brunt of injury. Nevertheless, this concept has not been formally incorporated into many of the current theories of disease pathogenesis, and it has not achieved a wide consensus. More importantly, in view of growing evidence that the molecular mechanisms of axon degeneration are separate and distinct from the canonical pathways of programmed cell death that mediate soma destruction, the possibility of early involvement of axons in PD has not been adequately emphasized as a rationale to explore the neurobiology of axons for novel therapeutic targets. We propose that ongoing degeneration of axons, not cell bodies, is the primary determinant of clinically apparent progression of disease, and that future experimental therapeutics intended to forestall disease progression will benefit from a new focus on the distinct mechanisms of axon degeneration.
Conflict of interest statement
Statement of Conflict: The authors have no financial interests to disclose
Figures
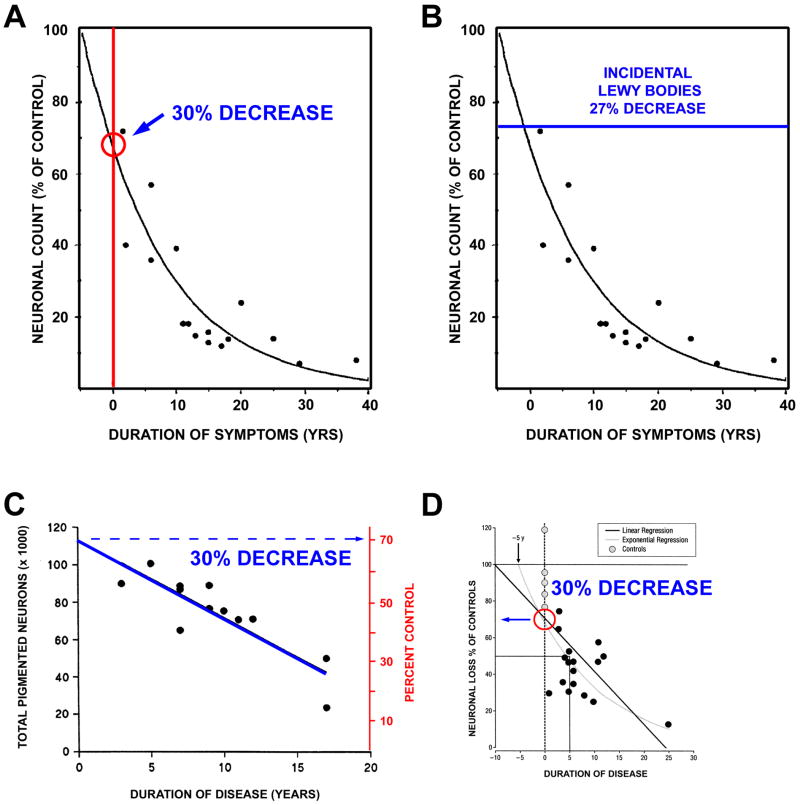
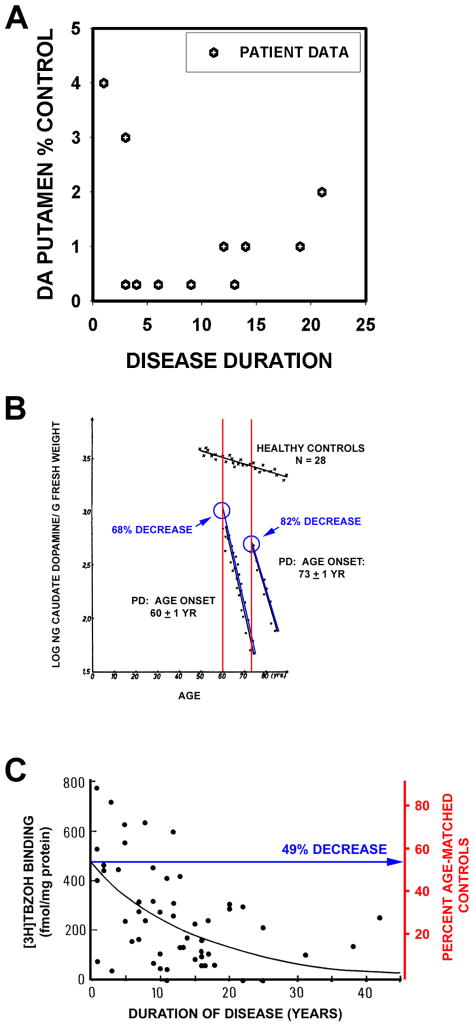
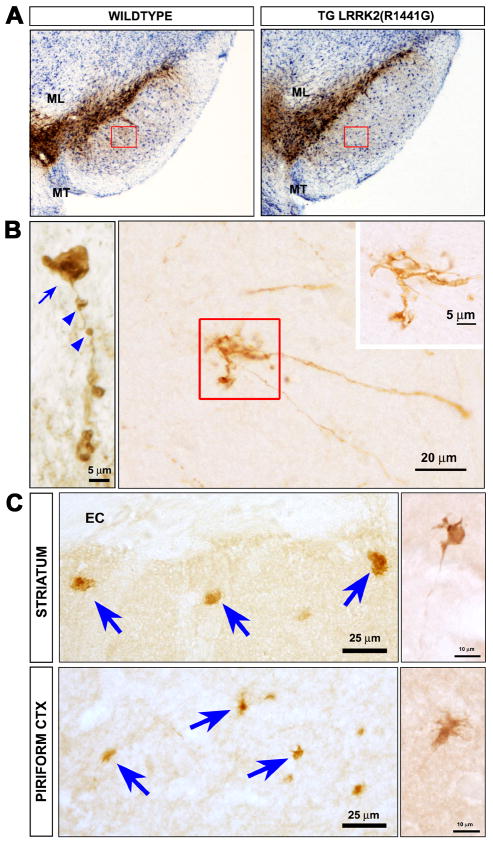
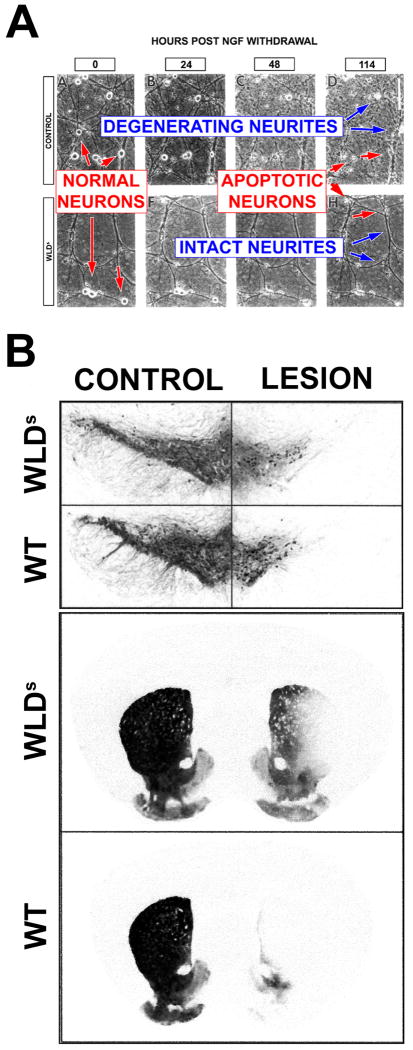
References
-
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. - PubMed
-
- Limousin P, Pollak P, Benazzouz A, et al. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995;345:91–95. - PubMed
-
- Shoulson I. Experimental therapeutics of neurodegenerative disorders: unmet needs. Science. 1998;282:1072–1074. - PubMed
-
- Olanow CW, Kieburtz K, Schapira AH. Why have we failed to achieve neuroprotection in Parkinson's disease? Ann Neurol. 2008;64 2:S101–S110. - PubMed
-
- Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annual Review of Cell Biology. 1991;7:663–698. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
