

The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses
- PMID: 20519382
- PMCID: PMC2916543
- DOI: 10.1128/JVI.02491-09
The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses
Abstract
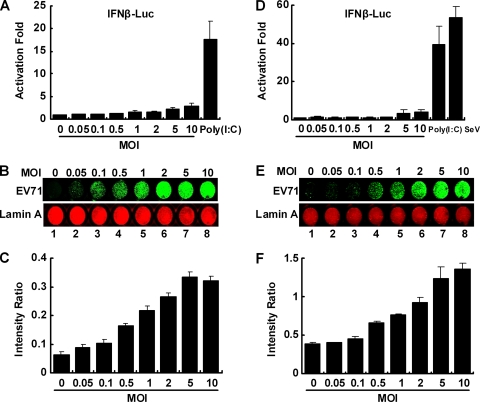
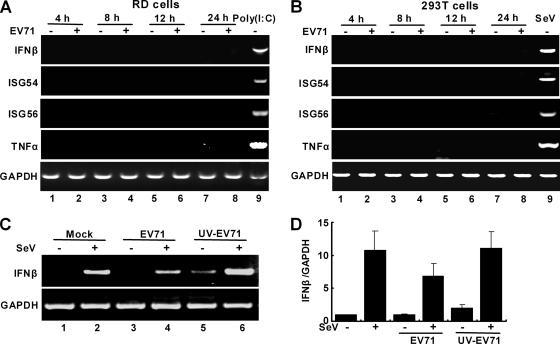
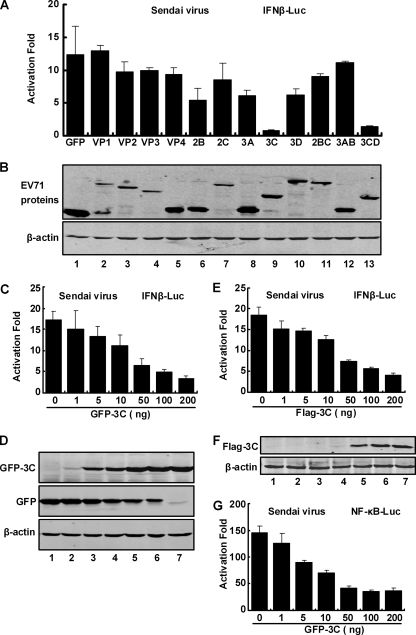
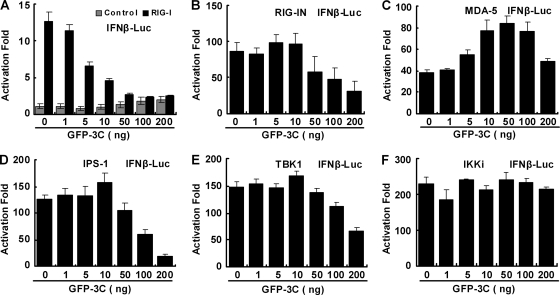
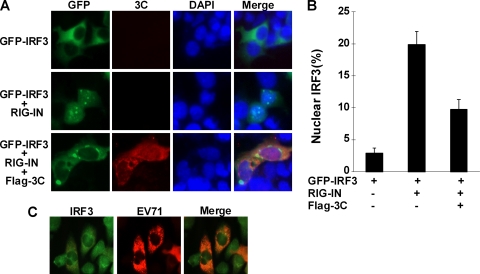
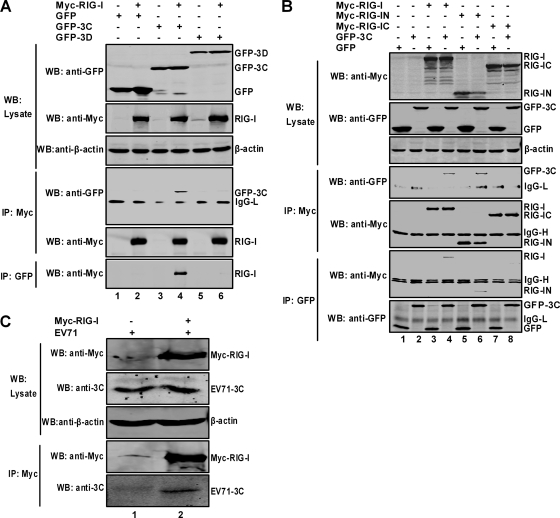
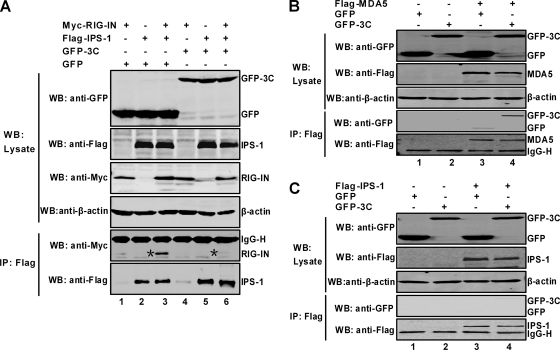
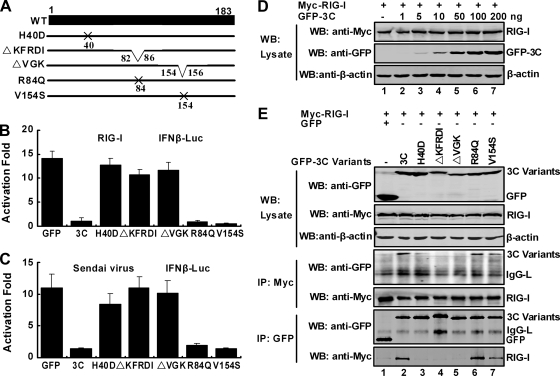
Enterovirus 71 (EV71) is a human pathogen that induces hand, foot, and mouth disease and fatal neurological diseases. Immature or impaired immunity is thought to associate with increased morbidity and mortality. In a murine model, EV71 does not facilitate the production of type I interferon (IFN) that plays a critical role in the first-line defense against viral infection. Administration of a neutralizing antibody to IFN-alpha/beta exacerbates the virus-induced disease. However, the molecular events governing this process remain elusive. Here, we report that EV71 suppresses the induction of antiviral immunity by targeting the cytosolic receptor retinoid acid-inducible gene I (RIG-I). In infected cells, EV71 inhibits the expression of IFN-beta, IFN-stimulated gene 54 (ISG54), ISG56, and tumor necrosis factor alpha. Among structural and nonstructural proteins encoded by EV71, the 3C protein is capable of inhibiting IFN-beta activation by virus and RIG-I. Nevertheless, EV71 3C exhibits no inhibitory activity on MDA5. Remarkably, when expressed in mammalian cells, EV71 3C associates with RIG-I via the caspase recruitment domain. This precludes the recruitment of an adaptor IPS-1 by RIG-I and subsequent nuclear translocation of interferon regulatory factor 3. An R84Q or V154S substitution in the RNA binding motifs has no effect. An H40D substitution is detrimental, but the protease activity associated with 3C is dispensable. Together, these results suggest that inhibition of RIG-I-mediated type I IFN responses by the 3C protein may contribute to the pathogenesis of EV71 infection.
Figures
Similar articles
-
Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3.J Virol. 2011 Sep;85(17):8811-8. doi: 10.1128/JVI.00447-11. Epub 2011 Jun 22. J Virol. 2011. PMID: 21697485 Free PMC article.
-
Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response.J Virol. 2014 Oct;88(19):11356-68. doi: 10.1128/JVI.01400-14. Epub 2014 Jul 23. J Virol. 2014. PMID: 25056901 Free PMC article.
-
Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses.J Virol. 2013 Feb;87(3):1690-8. doi: 10.1128/JVI.01855-12. Epub 2012 Nov 21. J Virol. 2013. PMID: 23175366 Free PMC article.
-
An update on enterovirus 71 infection and interferon type I response.Rev Med Virol. 2019 Jan;29(1):e2016. doi: 10.1002/rmv.2016. Epub 2018 Oct 30. Rev Med Virol. 2019. PMID: 30378208 Free PMC article. Review.
-
The tiers and dimensions of evasion of the type I interferon response by human cytomegalovirus.J Mol Biol. 2013 Dec 13;425(24):4857-71. doi: 10.1016/j.jmb.2013.08.023. Epub 2013 Sep 5. J Mol Biol. 2013. PMID: 24013068 Free PMC article. Review.
Cited by
-
Rotavirus nonstructural protein 1 antagonizes innate immune response by interacting with retinoic acid inducible gene I.Virol J. 2011 Dec 8;8:526. doi: 10.1186/1743-422X-8-526. Virol J. 2011. PMID: 22152002 Free PMC article.
-
Therapeutic and prevention strategies against human enterovirus 71 infection.World J Virol. 2015 May 12;4(2):78-95. doi: 10.5501/wjv.v4.i2.78. World J Virol. 2015. PMID: 25964873 Free PMC article. Review.
-
Apoptosis and Autophagy in Picornavirus Infection.Front Microbiol. 2019 Sep 3;10:2032. doi: 10.3389/fmicb.2019.02032. eCollection 2019. Front Microbiol. 2019. PMID: 31551969 Free PMC article. Review.
-
STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1.Int J Mol Sci. 2019 Sep 30;20(19):4870. doi: 10.3390/ijms20194870. Int J Mol Sci. 2019. PMID: 31575039 Free PMC article.
-
Immune Evasion of Enteroviruses Under Innate Immune Monitoring.Front Microbiol. 2018 Aug 14;9:1866. doi: 10.3389/fmicb.2018.01866. eCollection 2018. Front Microbiol. 2018. PMID: 30154774 Free PMC article. Review.
References
-
- AbuBakar, S., H. Y. Chee, M. F. Al-Kobaisi, J. Xiaoshan, K. B. Chua, and S. K. Lam. 1999. Identification of enterovirus 71 isolates from an outbreak of hand, foot and mouth disease (HFMD) with fatal cases of encephalomyelitis in Malaysia. Virus Res. 61:1-9. - PubMed
-
- Alexander, J. P., Jr., L. Baden, M. A. Pallansch, and L. J. Anderson. 1994. Enterovirus 71 infections and neurologic disease—United States, 1977-1991. J. Infect. Dis. 169:905-908. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
