

BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia patients on imatinib
- PMID: 20519627
- PMCID: PMC2995357
- DOI: 10.1182/blood-2008-10-183665
BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia patients on imatinib
Abstract
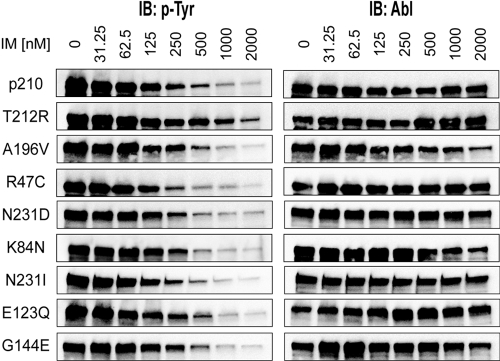
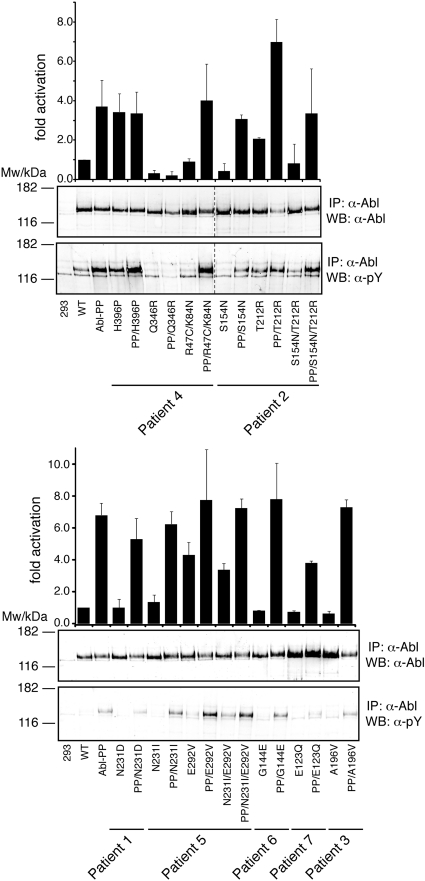
Point mutations in the kinase domain of BCR-ABL are the most common mechanism of drug resistance in chronic myeloid leukemia (CML) patients treated with ABL kinase inhibitors, including imatinib. It has also been shown in vitro that mutations outside the kinase domain in the neighboring linker, SH2, SH3, and Cap domains can confer imatinib resistance. In the context of ABL, these domains have an autoinhibitory effect on kinase activity, and mutations in this region can activate the enzyme. To determine the frequency and relevance to resistance of regulatory domain mutations in CML patients on imatinib, we screened for such mutations in a cohort of consecutive CML patients with various levels of response. Regulatory domain mutations were detected in 7 of 98 patients, whereas kinase domain mutations were detected in 29. One mutation (T212R) conferred in vitro tyrosine kinase inhibitor resistance and was associated with relapse, whereas most other mutations did not affect drug sensitivity. Mechanistic studies showed that T212R increased the activity of ABL and BCR-ABL and that T212R-induced resistance may be partially the result of stabilization of an active kinase conformation. Regulatory domain mutations are uncommon but may explain resistance in some patients without mutations in the kinase domain.
Figures




References
-
- Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. - PubMed
-
- Cortes J, O'Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004;104(7):2204–2205. - PubMed
-
- Higashi T, Tsukada J, Kato C, et al. Imatinib mesylate-sensitive blast crisis immediately after discontinuation of imatinib mesylate therapy in chronic myelogenous leukemia: report of two cases. Am J Hematol. 2004;76(3):275–278. - PubMed
-
- Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99(10):3530–3539. - PubMed
-
- Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99(9):3472–3475. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
