

Integrative genome comparison of primary and metastatic melanomas
- PMID: 20520718
- PMCID: PMC2875381
- DOI: 10.1371/journal.pone.0010770
Integrative genome comparison of primary and metastatic melanomas
Abstract
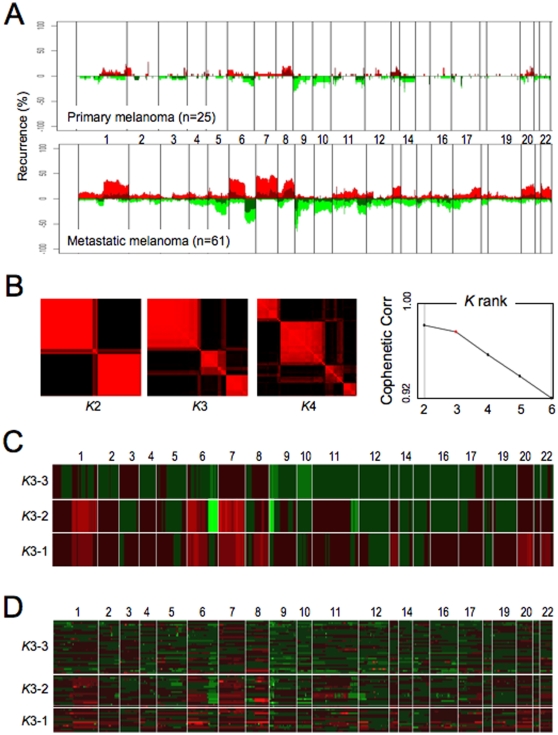
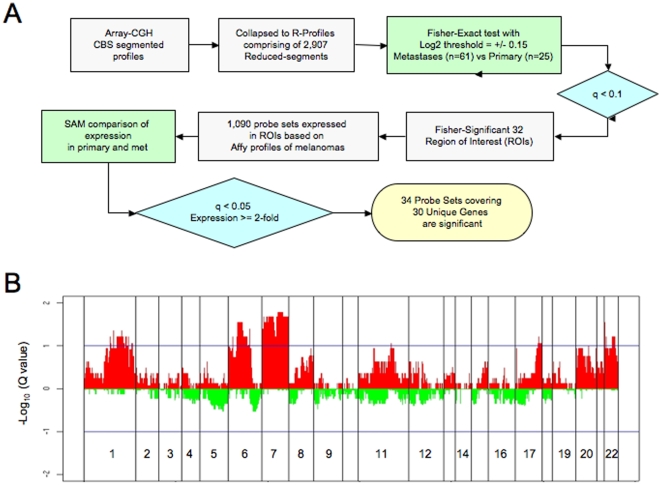
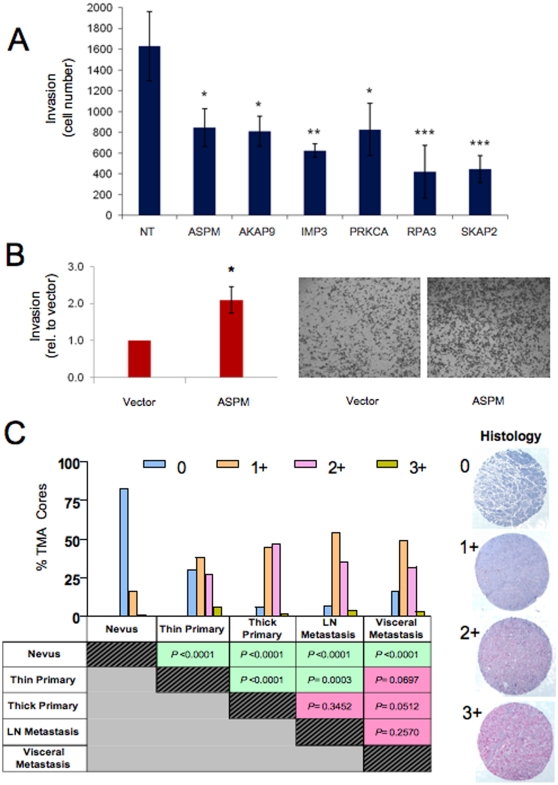
A cardinal feature of malignant melanoma is its metastatic propensity. An incomplete view of the genetic events driving metastatic progression has been a major barrier to rational development of effective therapeutics and prognostic diagnostics for melanoma patients. In this study, we conducted global genomic characterization of primary and metastatic melanomas to examine the genomic landscape associated with metastatic progression. In addition to uncovering three genomic subclasses of metastastic melanomas, we delineated 39 focal and recurrent regions of amplification and deletions, many of which encompassed resident genes that have not been implicated in cancer or metastasis. To identify progression-associated metastasis gene candidates, we applied a statistical approach, Integrative Genome Comparison (IGC), to define 32 genomic regions of interest that were significantly altered in metastatic relative to primary melanomas, encompassing 30 resident genes with statistically significant expression deregulation. Functional assays on a subset of these candidates, including MET, ASPM, AKAP9, IMP3, PRKCA, RPA3, and SCAP2, validated their pro-invasion activities in human melanoma cells. Validity of the IGC approach was further reinforced by tissue microarray analysis of Survivin showing significant increased protein expression in thick versus thin primary cutaneous melanomas, and a progression correlation with lymph node metastases. Together, these functional validation results and correlative analysis of human tissues support the thesis that integrated genomic and pathological analyses of staged melanomas provide a productive entry point for discovery of melanoma metastases genes.
Conflict of interest statement
Figures
References
-
- Tarhini AA, Agarwala SS. Cutaneous melanoma: available therapy for metastatic disease. Dermatol Ther. 2006;19:19–25. - PubMed
-
- Hoeflich KP, Herter S, Tien J, Wong L, Berry L, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–3051. - PubMed
-
- Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–2182. - PubMed
-
- Kim M, Gans JD, Nogueira C, Wang A, Paik JH, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
