

Prioritization of disease microRNAs through a human phenome-microRNAome network
- PMID: 20522252
- PMCID: PMC2880408
- DOI: 10.1186/1752-0509-4-S1-S2
Prioritization of disease microRNAs through a human phenome-microRNAome network
Abstract
Background: The identification of disease-related microRNAs is vital for understanding the pathogenesis of diseases at the molecular level, and is critical for designing specific molecular tools for diagnosis, treatment and prevention. Experimental identification of disease-related microRNAs poses considerable difficulties. Computational analysis of microRNA-disease associations is an important complementary means for prioritizing microRNAs for further experimental examination.
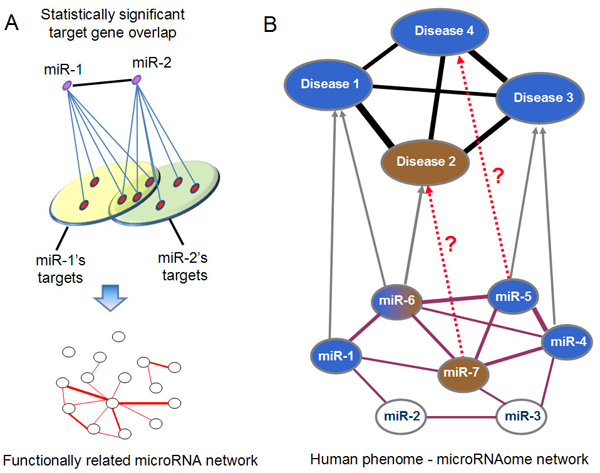
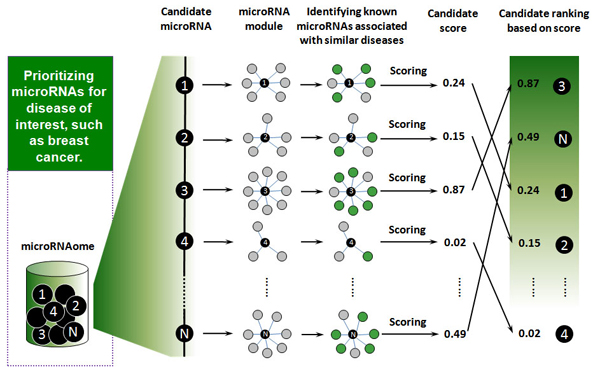
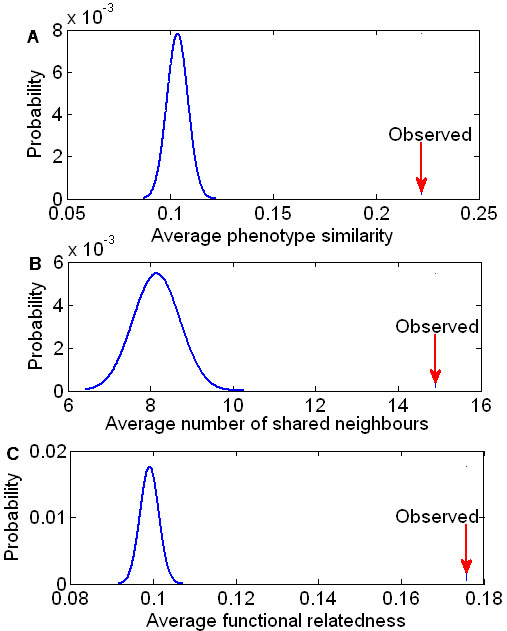
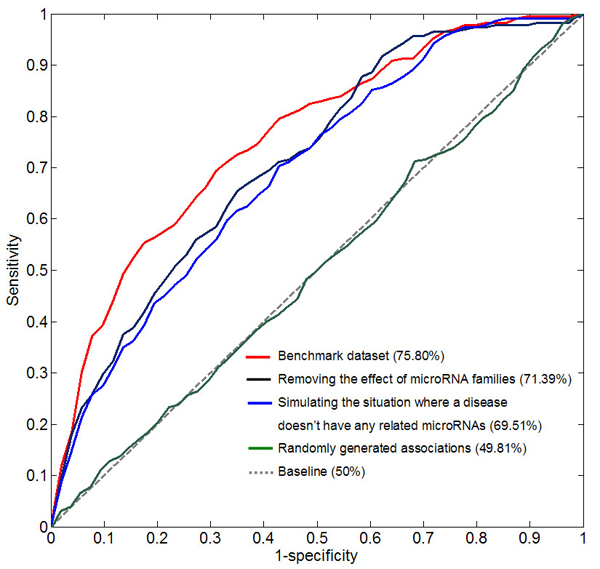
Results: Herein, we devised a computational model to infer potential microRNA-disease associations by prioritizing the entire human microRNAome for diseases of interest. We tested the model on 270 known experimentally verified microRNA-disease associations and achieved an area under the ROC curve of 75.80%. Moreover, we demonstrated that the model is applicable to diseases with which no known microRNAs are associated. The microRNAome-wide prioritization of microRNAs for 1,599 disease phenotypes is publicly released to facilitate future identification of disease-related microRNAs.
Conclusions: We presented a network-based approach that can infer potential microRNA-disease associations and drive testable hypotheses for the experimental efforts to identify the roles of microRNAs in human diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
