

Beyond rotamers: a generative, probabilistic model of side chains in proteins
- PMID: 20525384
- PMCID: PMC2902450
- DOI: 10.1186/1471-2105-11-306
Beyond rotamers: a generative, probabilistic model of side chains in proteins
Abstract
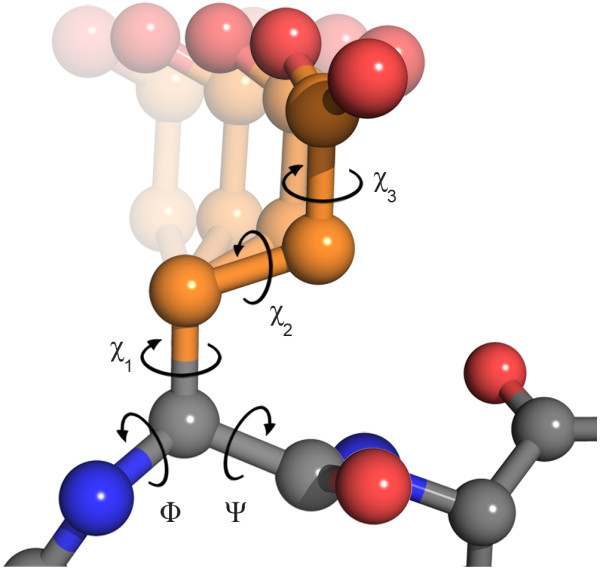
Background: Accurately covering the conformational space of amino acid side chains is essential for important applications such as protein design, docking and high resolution structure prediction. Today, the most common way to capture this conformational space is through rotamer libraries - discrete collections of side chain conformations derived from experimentally determined protein structures. The discretization can be exploited to efficiently search the conformational space. However, discretizing this naturally continuous space comes at the cost of losing detailed information that is crucial for certain applications. For example, rigorously combining rotamers with physical force fields is associated with numerous problems.
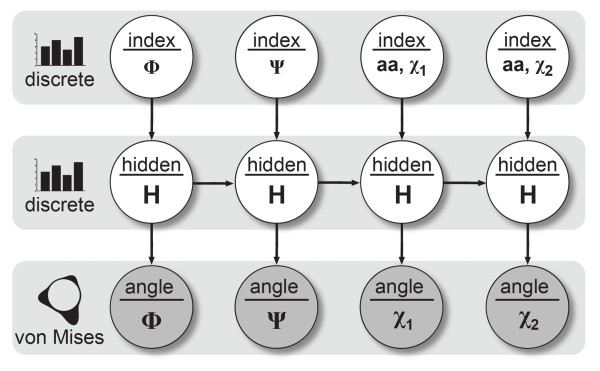
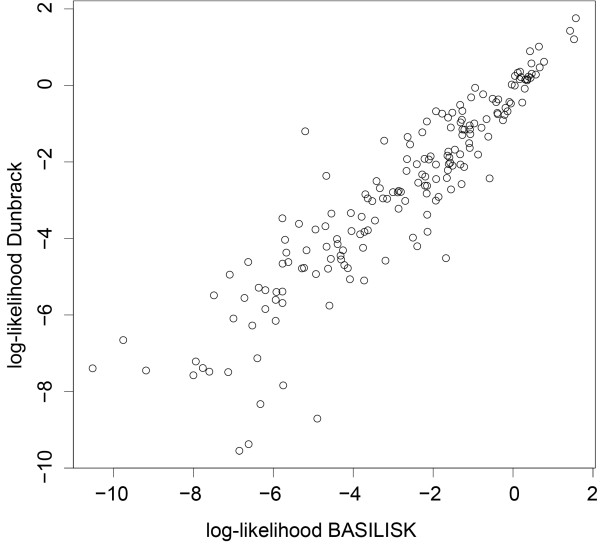
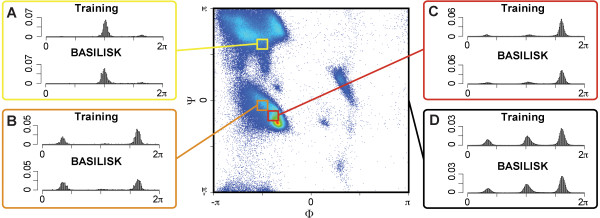
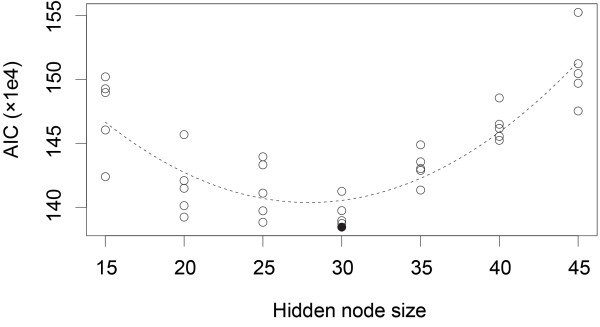
Results: In this work we present BASILISK: a generative, probabilistic model of the conformational space of side chains that makes it possible to sample in continuous space. In addition, sampling can be conditional upon the protein's detailed backbone conformation, again in continuous space - without involving discretization.
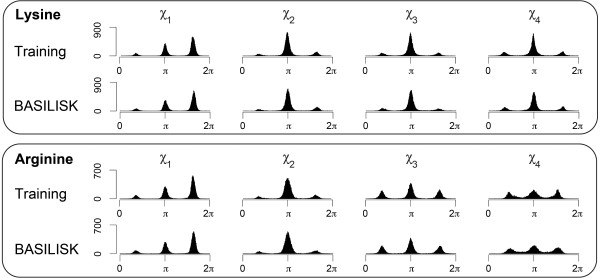
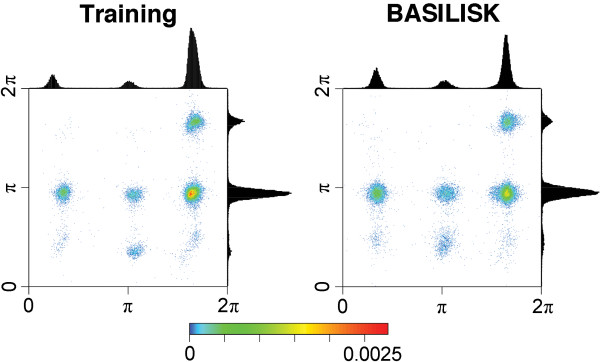
Conclusions: A careful analysis of the model and a comparison with various rotamer libraries indicates that the model forms an excellent, fully continuous model of side chain conformational space. We also illustrate how the model can be used for rigorous, unbiased sampling with a physical force field, and how it improves side chain prediction when used as a pseudo-energy term. In conclusion, BASILISK is an important step forward on the way to a rigorous probabilistic description of protein structure in continuous space and in atomic detail.
Figures
Similar articles
-
Protein design using continuous rotamers.PLoS Comput Biol. 2012 Jan;8(1):e1002335. doi: 10.1371/journal.pcbi.1002335. Epub 2012 Jan 12. PLoS Comput Biol. 2012. PMID: 22279426 Free PMC article.
-
Toward the Accuracy and Speed of Protein Side-Chain Packing: A Systematic Study on Rotamer Libraries.J Chem Inf Model. 2020 Jan 27;60(1):410-420. doi: 10.1021/acs.jcim.9b00812. Epub 2019 Dec 31. J Chem Inf Model. 2020. PMID: 31851497 Free PMC article.
-
Rotamer libraries and probabilities of transition between rotamers for the side chains in protein-protein binding.Proteins. 2012 Aug;80(8):2089-98. doi: 10.1002/prot.24103. Epub 2012 Jun 12. Proteins. 2012. PMID: 22544766 Free PMC article.
-
Rotamer Dynamics: Analysis of Rotamers in Molecular Dynamics Simulations of Proteins.Biophys J. 2019 Jun 4;116(11):2062-2072. doi: 10.1016/j.bpj.2019.04.017. Epub 2019 Apr 22. Biophys J. 2019. PMID: 31084902 Free PMC article. Review.
-
Sifting through the noise: A survey of diffusion probabilistic models and their applications to biomolecules.J Mol Biol. 2025 Mar 15;437(6):168818. doi: 10.1016/j.jmb.2024.168818. Epub 2024 Oct 9. J Mol Biol. 2025. PMID: 39389290 Review.
Cited by
-
To pack or not to pack: revisiting protein side-chain packing in the post-AlphaFold era.bioRxiv [Preprint]. 2025 Feb 27:2025.02.22.639681. doi: 10.1101/2025.02.22.639681. bioRxiv. 2025. Update in: Brief Bioinform. 2025 May 1;26(3):bbaf297. doi: 10.1093/bib/bbaf297. PMID: 40060396 Free PMC article. Updated. Preprint.
-
UniCon3D: de novo protein structure prediction using united-residue conformational search via stepwise, probabilistic sampling.Bioinformatics. 2016 Sep 15;32(18):2791-9. doi: 10.1093/bioinformatics/btw316. Epub 2016 Jun 3. Bioinformatics. 2016. PMID: 27259540 Free PMC article.
-
Protein design using continuous rotamers.PLoS Comput Biol. 2012 Jan;8(1):e1002335. doi: 10.1371/journal.pcbi.1002335. Epub 2012 Jan 12. PLoS Comput Biol. 2012. PMID: 22279426 Free PMC article.
-
FragBuilder: an efficient Python library to setup quantum chemistry calculations on peptides models.PeerJ. 2014 Mar 4;2:e277. doi: 10.7717/peerj.277. eCollection 2014. PeerJ. 2014. PMID: 24688855 Free PMC article.
-
ProCS15: a DFT-based chemical shift predictor for backbone and Cβ atoms in proteins.PeerJ. 2015 Oct 20;3:e1344. doi: 10.7717/peerj.1344. eCollection 2015. PeerJ. 2015. PMID: 26623185 Free PMC article.
References
-
- Chandrasekaran R, Ramachandran GN. Studies on the conformation of amino acids. XI. Analysis of the observed side group conformation in proteins. Int J Protein Res. 1970;2:223–233. - PubMed
-
- Eyring H. Steric hindrance and collision diameters. J Am Chem Soc. 1932;54:3191–3203. doi: 10.1021/ja01347a022. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
