

Properties of consensus methods for inferring species trees from gene trees
- PMID: 20525567
- PMCID: PMC2909780
- DOI: 10.1093/sysbio/syp008
Properties of consensus methods for inferring species trees from gene trees
Abstract
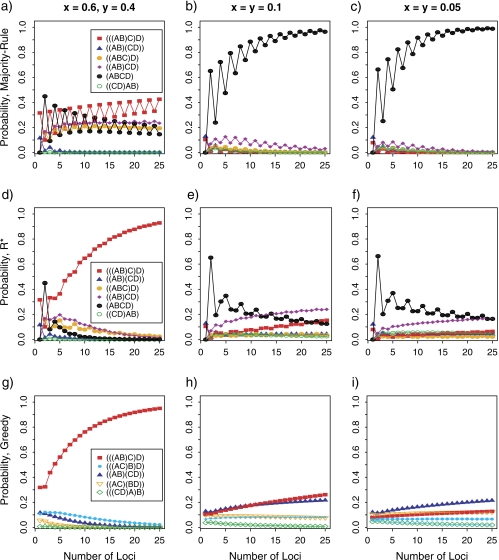
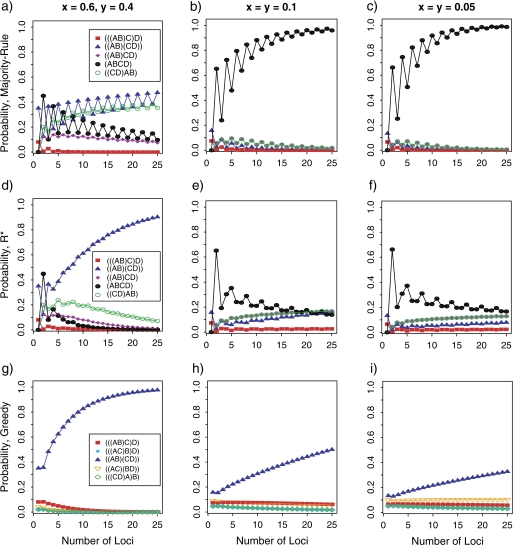
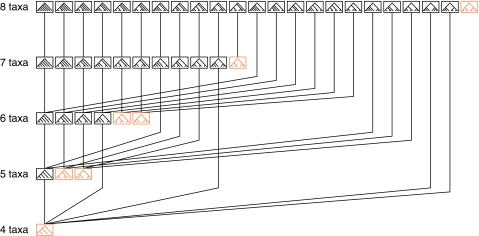
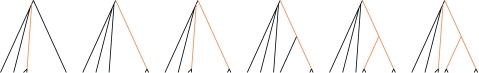
Consensus methods provide a useful strategy for summarizing information from a collection of gene trees. An important application of consensus methods is to combine gene trees to estimate a species tree. To investigate the theoretical properties of consensus trees that would be obtained from large numbers of loci evolving according to a basic evolutionary model, we construct consensus trees from rooted gene trees that occur in proportion to gene-tree probabilities derived from coalescent theory. We consider majority-rule, rooted triple (R(*)), and greedy consensus trees obtained from known, rooted gene trees, both in the asymptotic case as numbers of gene trees approach infinity and for finite numbers of genes. Our results show that for some combinations of species-tree branch lengths, increasing the number of independent loci can make the rooted majority-rule consensus tree more likely to be at least partially unresolved. However, the probability that the R(*) consensus tree has the species-tree topology approaches 1 as the number of gene trees approaches infinity. Although the greedy consensus algorithm can be the quickest to converge on the correct species-tree topology when increasing the number of gene trees, it can also be positively misleading. The majority-rule consensus tree is not a misleading estimator of the species-tree topology, and the R(*) consensus tree is a statistically consistent estimator of the species-tree topology. Our results therefore suggest a method for using multiple loci to infer the species-tree topology, even when it is discordant with the most likely gene tree.
Figures
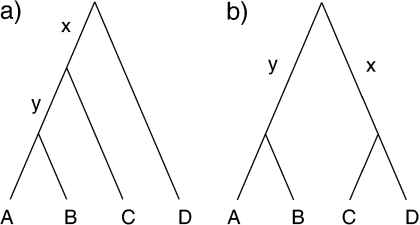
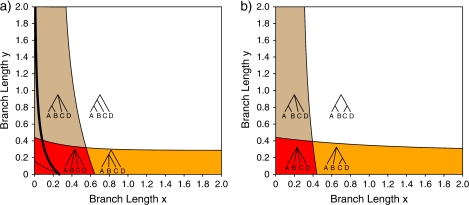
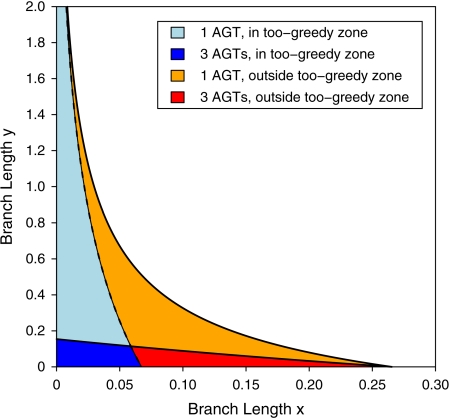
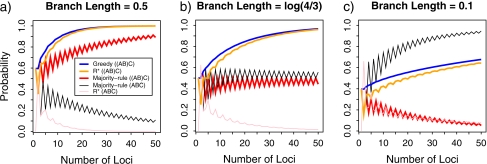
References
-
- Ané C, Larget B, Baum DA, Smith SD, Rokas A. Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 2007;24:412–426. - PubMed
-
- Baum BR. Combining trees as a way of combining data sets for phylogenetic inference, and the desirability of combining trees. Taxon. 1992;41:3–10.
-
- Baum DA. Concordance trees, concordance factors, and the exploration of reticulate genealogy. Taxon. 2007;56:417–426.
-
- Bremer K. Combinable component consensus. Cladistics. 1990;6:369–372. - PubMed
-
- Bryant D. A classification of consensus methods for phylogenies. In: Janowitz M, Lapointe F-J, McMorris FR, Mirkin B, Roberts FS, editors. BioConsensus. Providence (RI): Center for Discrete Mathematics and Theoretical Computer Science, American Mathematical Society; 2003. pp. 163–183.
